Медицински експерт на статията

Нови публикации
Наследствен нефрит (синдром на Алпорт) при деца
Последно прегледани: 05.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
Наследственият нефрит (синдром на Алпорт) е генетично обусловена наследствена неимунна гломерулопатия, проявяваща се с хематурия (понякога с протеинурия), прогресивно намаляване на бъбречната функция с развитие на хронична бъбречна недостатъчност, често комбинирана със сензоневрална глухота и зрителни увреждания.
Заболяването е описано за първи път през 1902 г. от Л. Г. Гътри, който наблюдава семейство, в което хематурия се наблюдава в няколко поколения. През 1915 г. А. Ф. Хърст описва развитието на уремия при членове на същото семейство. През 1927 г. А. Алпорт за първи път идентифицира загуба на слуха при няколко роднини с хематурия. През 50-те години на миналия век са описани очни лезии при подобно заболяване. През 1972 г. при пациенти с наследствена хематурия, по време на морфологично изследване на бъбречната тъкан, Хинглайс и др. разкриват неравномерно разширяване и стратификация на гломерулните базални мембрани. През 1985 г. е идентифицирана генетичната основа на наследствения нефрит - мутация в гена за колаген тип IV (Fiengold et al., 1985).
Изследването на генетичната природа на заболяването ни позволи да заключим, че разликите във фенотипните прояви на наследствения нефрит (със или без загуба на слуха) се дължат на степента на експресия на мутантния ген. По този начин, понастоящем всички клинични варианти се считат за прояви на едно заболяване и терминът „наследствен нефрит“ е синоним на термина „синдром на Алпорт“.
Според епидемиологични проучвания, наследственият нефрит се среща с честота 17 на 100 000 деца.
 [
[ Причини за синдром на Алпорт
Генетичната основа на заболяването е мутация в гена на а-5 веригата на колаген тип IV. Този тип е универсален за базалните мембрани на бъбреците, кохлеарния апарат, капсулата на лещата, ретината и роговицата на окото, което е доказано в изследвания, използващи моноклонални антитела срещу тази колагенова фракция. Наскоро е посочена възможността за използване на ДНК сонди за пренатална диагностика на наследствен нефрит.
Подчертава се значението на тестването на всички членове на семейството с ДНК сонди за идентифициране на носители на мутантния ген, което е от голямо значение при провеждането на медико-генетично консултиране на семейства с това заболяване. Въпреки това, до 20% от семействата нямат роднини, страдащи от бъбречно заболяване, което предполага висока честота на спонтанни мутации на анормалния ген. Повечето пациенти с наследствен нефрит имат в семействата си лица с бъбречно заболяване, загуба на слуха и зрителна патология; кръвните бракове между хора с един или повече предци са важни, тъй като при брака на роднини вероятността за получаване на едни и същи гени от двамата родители се увеличава. Установени са автозомно доминантни, автозомно рецесивни и доминантни, Х-свързани пътища на предаване.
При децата най-често се разграничават три вида наследствен нефрит: синдром на Алпорт, наследствен нефрит без загуба на слуха и фамилна доброкачествена хематурия.
Синдромът на Алпорт е наследствен нефрит с увреждане на слуха. В основата му е комбиниран дефект в структурата на колагена на гломерулната базална мембрана на бъбреците, ушните и очните структури. Генът на класическия синдром на Алпорт се намира в локус 21-22 q на дългото рамо на Х хромозомата. В повечето случаи се унаследява по доминантен начин, свързан с Х хромозомата. В тази връзка синдромът на Алпорт е по-тежък при мъжете, тъй като при жените функцията на мутантния ген се компенсира от здрав алел на втората, неувредена хромозома.
Генетичната основа за развитието на наследствен нефрит са мутации в гените на алфа веригите на колаген тип IV. Известни са шест алфа вериги на колаген тип IV G: гените на a5- и a6-веригите (Col4A5 и Col4A5) са разположени на дългото рамо на X хромозомата в зоната 21-22q; гените на a3- и a4-веригите (Col4A3 и Col4A4) са на втората хромозома; гените на a1- и a2-веригите (Col4A1 и Col4A2) са на 13-тата хромозома.
В повечето случаи (80-85%) се открива Х-свързан модел на унаследяване на заболяването, свързан с увреждане на гена Col4A5 в резултат на делеция, точкови мутации или нарушения на сплайсинга. В момента са открити повече от 200 мутации на гена Col4A5, отговорни за нарушаването на синтеза на a5-веригите на колаген тип IV. При този тип унаследяване заболяването се проявява при деца от двата пола, но при момчетата е по-тежко.
Мутациите в локусите на гените Col4A3 и Col4A4, отговорни за синтеза на веригите a3 и a4 на колаген тип IV, се унаследяват автозомно. Според изследвания, автозомно доминантният тип наследяване се наблюдава в 16% от случаите на наследствен нефрит, а автозомно рецесивен тип - при 6% от пациентите. Известни са около 10 варианта на мутации на гените Col4A3 и Col4A4.
Резултатът от мутациите е нарушение на процесите на сглобяване на колаген тип IV, което води до нарушаване на неговата структура. Колагенът тип IV е един от основните компоненти на гломерулната базална мембрана, кохлеарния апарат и лещата на окото, чиято патология ще бъде открита в клиниката на наследствения нефрит.
Колаген тип IV, който е част от гломерулната базална мембрана, се състои главно от две a1-вериги (IV) и една a2-верига (IV), а също така съдържа a3, a4, a5-вериги. Най-често при Х-свързано наследяване мутацията на гена Col4A5 е съпроводена с отсъствие на a3-, a4-, a5- и a6-вериги в структурата на колаген тип IV, а броят на o1- и a2-веригите в гломерулната базална мембрана се увеличава. Механизмът на това явление е неясен, предполага се, че причината са посттранскрипционни промени в mRNA.
Липсата на a3, a4 и a5 вериги в структурата на колаген тип IV на гломерулните базални мембрани води до тяхното изтъняване и чупливост в ранните стадии на синдрома на Алпорт, което клинично се проявява по-често с хематурия (по-рядко с хематурия с протеинурия или само с протеинурия), загуба на слуха и лентиконус. По-нататъшното прогресиране на заболяването води до удебеляване и нарушена пропускливост на базалните мембрани в късните стадии на заболяването, с пролиферация на колаген тип V и VI в тях, проявяваща се в повишаване на протеинурията и намаляване на бъбречната функция.
Характерът на мутацията, лежаща в основата на наследствения нефрит, до голяма степен определя неговата фенотипна проява. В случай на делеция на Х хромозомата с едновременна мутация на гените Col4A5 и Col4A6, отговорни за синтеза на a5- и a6-веригите на колаген тип IV, синдромът на Алпорт се комбинира с лейомиоматоза на хранопровода и гениталиите. Според данни от изследвания, в случай на мутация на гена Col4A5, свързана с делеция, се отбелязва по-голяма тежест на патологичния процес, комбинация от бъбречно увреждане с екстраренални прояви и ранно развитие на хронична бъбречна недостатъчност, в сравнение с точкова мутация на този ген.
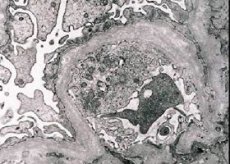
Морфологично, електронната микроскопия разкрива изтъняване и стратификация на гломерулните базални мембрани (особено lamina densa) и наличие на електронно-плътни гранули. Гломерулните лезии могат да бъдат хетерогенни при един и същ пациент, от минимални фокални мезангиални лезии до гломерулосклероза. Гломерулитът при синдром на Алпорт винаги е имунонегативен, което го отличава от гломерулонефрита. Характерните особености включват развитие на тубулна атрофия, лимфохистиоцитна инфилтрация и наличие на "пенести клетки" с липидни включвания - липофаги. С напредването на заболяването се разкриват удебеляване и изразено разрушаване на гломерулните базални мембрани.
Разкриват се определени промени в имунната система. Пациентите с наследствен нефрит имат понижено ниво на Ig A и склонност към повишаване на концентрацията на IgM в кръвта, като нивото на IgG може да е повишено в ранните стадии на заболяването и да намалява в по-късните. Вероятно повишаването на концентрацията на IgM и G е вид компенсаторна реакция в отговор на дефицит на IgA.
Функционалната активност на Т-лимфоцитната система е намалена; отбелязва селективно намаляване на В-лимфоцитите, отговорни за синтеза на Ig A, нарушава се фагоцитната връзка на имунитета, главно поради нарушаване на хемотаксиса и вътреклетъчните процеси на храносмилане в неутрофилите.
При изследване на бъбречна биопсия при пациенти със синдром на Алпорт, данните от електронната микроскопия разкриват ултраструктурни промени в гломерулната базална мембрана: изтъняване, нарушаване на структурата и разцепване на гломерулните базални мембрани с промяна в дебелината им и неравномерни контури. В ранните стадии на наследствен нефрит дефектът определя изтъняването и чупливостта на гломерулните базални мембрани.
Изтъняването на гломерулните мембрани е по-благоприятен признак и е по-често срещано при момичетата. По-постоянен електронно-микроскопски признак при наследствен нефрит е разцепването на базалната мембрана, като тежестта на нейното разрушаване корелира с тежестта на процеса.
Симптоми на синдрома на Алпорт при деца
Първите симптоми на синдрома на Алпорт под формата на изолиран уринарен синдром най-често се откриват при деца през първите три години от живота. В повечето случаи заболяването се открива случайно. Уринарният синдром се открива по време на профилактичен преглед на детето, преди приемане в детско заведение или по време на остри респираторни инфекции (ОРВИ). В случай на патология в урината по време на ОРВИ. При наследствен нефрит, за разлика от придобития гломерулонефрит, няма латентен период.
В началния стадий на заболяването здравето на детето страда малко, характерна черта е персистирането и резистентността на уринарния синдром. Един от основните признаци е хематурия с различна степен на тежест, наблюдавана в 100% от случаите. Увеличаване на степента на хематурия се отбелязва по време или след респираторни инфекции, физическа активност или след превантивни ваксинации. Протеинурията в повечето случаи не надвишава 1 g/ден, в началото на заболяването може да бъде непостоянна, с напредването на процеса протеинурията се увеличава. Периодично в уринарния седимент може да се наблюдава левкоцитурия с преобладаване на лимфоцити, което е свързано с развитието на интерстициални промени.
Впоследствие се нарушава частична бъбречна функция, общото състояние на пациента се влошава: появяват се интоксикация, мускулна слабост, артериална хипотония, често се появяват нарушения на слуха (особено при момчетата), а понякога и зрителни нарушения. Интоксикацията се проявява с бледност, умора и главоболие. В началния стадий на заболяването загубата на слуха в повечето случаи се открива само чрез аудиография. Загубата на слуха при синдром на Алпорт може да се появи в различни периоди от детството, но най-често загубата на слуха се диагностицира на възраст 6-10 години. Загубата на слуха при децата започва с високи честоти, достигайки значителна степен във въздушната и костната проводимост, преминавайки от звукопроводяща към звуковъзприемаща загуба на слуха. Загубата на слуха може да бъде един от първите симптоми на заболяването и може да предшества пикочния синдром.
В 20% от случаите пациентите със синдром на Алпорт имат промени в зрителните органи. Най-често откриваните аномалии са тези на лещата: сферофокия, преден, заден или смесен лентиконус и различни катаракти. В семейства със синдром на Алпорт има значителна честота на миопия. Редица изследователи постоянно отбелязват двустранни перимакуларни промени в тези семейства под формата на ярки белезникави или жълтеникави гранулации в жълтото тяло. Те считат този признак за постоянен симптом, който има висока диагностична стойност при синдрома на Алпорт. KS Chugh et al. (1993) в офталмологично проучване установяват при пациенти със синдром на Алпорт намаление на зрителната острота в 66,7% от случаите, преден лентиконус в 37,8%, петна по ретината в 22,2%, катаракта в 20% и кератоконус в 6,7%.
При някои деца с наследствен нефрит, особено при развитие на бъбречна недостатъчност, се наблюдава значително изоставане във физическото развитие. С прогресирането на бъбречната недостатъчност се развива артериална хипертония. При децата тя се открива по-често в юношеска възраст и в по-възрастни възрастови групи.
Пациентите с наследствен нефрит се характеризират с наличието на различни (повече от 5-7) стигми на съединителнотъканна дисморфогенеза. Сред стигмите на съединителнотъканната тъкан при пациентите най-често срещани са хипертелоризъм на очите, високо небце, аномалии на захапката, анормална форма на ушните миди, изкривяване на малкия пръст на ръцете и "сандалово празнина" на краката. Наследственият нефрит се характеризира с еднородност на стигмите на дисморфогенезата в рамките на едно семейство, както и с висока честота на разпространението им сред роднини на пробанди, по чиято линия се предава заболяването.
В ранните стадии на заболяването се установява изолирано намаляване на парциалните бъбречни функции: транспорт на аминокиселини, електролити, концентрационна функция, ацидогенеза, по-късните промени засягат функционалното състояние както на проксималните, така и на дисталните части на нефрона и се характеризират с комбинирани парциални нарушения. Намаляване на гломерулната филтрация настъпва по-късно, по-често в юношеска възраст. С прогресирането на наследствения нефрит се развива анемия.
По този начин, наследственият нефрит се характеризира с поетапно протичане на заболяването: първо, латентен стадий или скрити клинични симптоми, проявяващи се с минимални промени в пикочния синдром, след което настъпва постепенна декомпенсация на процеса с намаляване на бъбречната функция с изявени клинични симптоми (интоксикация, астения, забавяне на развитието, анемия). Клиничните симптоми обикновено се появяват независимо от наслояването на възпалителната реакция.
Наследственият нефрит може да се прояви в различни възрастови периоди, което зависи от действието на гена, който е в потиснато състояние до определен момент.
Класификация
Има три вида наследствен нефрит
- Вариант I - клинично се проявява като нефрит с хематурия, загуба на слуха и увреждане на очите. Ходът на нефрита е прогресиращ с развитието на хронична бъбречна недостатъчност. Типът на унаследяване е доминантен, свързан с Х хромозомата. Морфологично се разкрива нарушение на структурата на базалната мембрана, нейното изтъняване и разцепване.
- Вариант II - клинично се проявява като нефрит с хематурия без загуба на слуха. Ходът на нефрита е прогресиращ с развитието на хронична бъбречна недостатъчност. Типът на унаследяване е доминантен, свързан с Х хромозомата. Морфологично се установява изтъняване на гломерулно-капилярната базална мембрана (особено ламинаденса).
- Вариант III - доброкачествена фамилна хематурия. Ходът е благоприятен, хронична бъбречна недостатъчност не се развива. Типът на унаследяване е автозомно доминантен или автозомно рецесивен. При автозомно рецесивен тип наследяване се наблюдава по-тежко протичане на заболяването при жените.
Диагностика на синдрома на Алпорт
Предлагат се следните критерии:
- наличието на поне двама пациенти с нефропатия във всяко семейство;
- хематурия като водещ симптом на нефропатия при пробанда;
- наличие на загуба на слуха при поне един член на семейството;
- развитие на хронична бъбречна недостатъчност при един или повече роднини.
В диагностиката на различни наследствени и вродени заболявания голямо място се отделя на цялостния подход към изследването и най-вече на вниманието към данните, получени при съставяне на родословието на детето. Диагнозата синдром на Алпорт се счита за валидна в случаите, когато при пациента се откриват 3 от 4 типични признака: наличие на хематурия и хронична бъбречна недостатъчност в семейството, наличие на невросензорна загуба на слуха, патология на зрението при пациента, откриване на признаци на разцепване на гломерулната базална мембрана с промяна в нейната дебелина и неравни контури по време на електронно-микроскопски характеристики на биопсията.
Прегледът на пациента трябва да включва клинични и генетични методи на изследване; целенасочено проучване на историята на заболяването; общ преглед на пациента, като се вземат предвид диагностично значимите критерии. В стадия на компенсация патологията може да бъде открита само чрез фокусиране върху синдроми като наличие на наследствена обремененост, хипотония, множество стигми на дизембриогенезата, промени в уринарния синдром. В стадия на декомпенсация могат да се появят екстраренални симптоми, като тежка интоксикация, астения, забавено физическо развитие, анемия, проявяващи се и засилващи се с постепенно намаляване на бъбречната функция. При повечето пациенти с намаляване на бъбречната функция се наблюдава следното: намалена ацидо- и аминогенеза; 50% от пациентите отбелязват значително намаляване на секреторната функция на бъбреците; ограничен диапазон на колебания в оптичната плътност на урината; нарушение на филтрационния ритъм и след това намаляване на гломерулната филтрация. Стадият на хронична бъбречна недостатъчност се диагностицира, когато пациентите имат повишено ниво на урея в кръвния серум (повече от 0,35 g/l) в продължение на 3-6 месеца или повече и намаление на гломерулната филтрация до 25% от нормата.
Диференциалната диагностика на наследствения нефрит трябва да се провежда предимно с хематуричната форма на придобит гломерулонефрит. Придобитият гломерулонефрит най-често има остро начало, период от 2-3 седмици след инфекция, екстраренални признаци, включително хипертония от първите дни (при наследствения нефрит, напротив, хипотония), намалена гломерулна филтрация в началото на заболяването, липса на нарушение на частичните тубулни функции, докато при наследствения те са налице. Придобитият гломерулонефрит протича с по-изразена хематурия и протеинурия, с повишена СУЕ. Типичните промени в гломерулната базална мембрана, характерни за наследствения нефрит, са с диагностична стойност.
Диференциалната диагноза от дисметаболитна нефропатия се провежда с хронична бъбречна недостатъчност, в семейството клинично разкрити хетерогенни бъбречни заболявания, като може да има спектър от нефропатия от пиелонефрит до уролитиаза. Децата често имат оплаквания от болка в корема и периодично по време на уриниране, в утайката на урината - оксалати.
При съмнение за наследствен нефрит, пациентът трябва да бъде насочен към специализирано нефрологично отделение за уточняване на диагнозата.
Какво трябва да проучим?
Какви тестове са необходими?
Към кого да се свържете?
Лечение на синдрома на Алпорт
Режимът включва ограничения за тежки физически натоварвания и престой на чист въздух. Диетата е пълноценна, с достатъчни нива на пълноценни протеини, мазнини и въглехидрати, като се отчита бъбречната функция. От голямо значение е откриването и лечението на хронични огнища на инфекция. Използват се следните медикаменти: АТФ, кокарбоксилаза, пиридоксин (до 50 мг/ден), карнитин хлорид. Курсовете се прилагат 2-3 пъти годишно. При хематурия се предписва билково лекарство - коприва, сок от арония, бял равнец.
В чуждестранната и местната литература има съобщения за лечение с преднизолон и употребата на цитостатици. Трудно е обаче да се прецени ефектът.
При хронична бъбречна недостатъчност се използват хемодиализа и бъбречна трансплантация.
Няма методи за специфична (ефективна патогенетична) терапия за наследствен нефрит. Всички лечебни мерки са насочени към предотвратяване и забавяне на намаляването на бъбречната функция.
Диетата трябва да бъде балансирана и висококалорична, като се отчита функционалното състояние на бъбреците. При липса на функционални нарушения, диетата на детето трябва да съдържа достатъчно количество протеини, мазнини и въглехидрати. При наличие на признаци на бъбречна дисфункция, количеството протеини, въглехидрати, калций и фосфор трябва да се ограничи, което забавя развитието на хронична бъбречна недостатъчност.
Физическата активност трябва да бъде ограничена; на децата се препоръчва да избягват спорт.
Трябва да се избягва контакт с инфекциозно болни, да се намали рискът от развитие на остри респираторни заболявания. Необходима е санация на огнищата на хронична инфекция. Превантивни ваксинации не се провеждат за деца с наследствен нефрит, ваксинацията е възможна само по епидемиологични показания.
Хормоналната и имуносупресивна терапия при наследствен нефрит е неефективна. Има индикации за известен положителен ефект (намаляване на протеинурията и забавяне на прогресията на заболяването) при продължителна многогодишна употреба на циклоспорин А и АСЕ инхибитори.
При лечението на пациенти се използват лекарства, които подобряват метаболизма:
- пиридоксин - 2-3 мг/кг/ден в 3 дози в продължение на 4 седмици;
- кокарбоксилаза - 50 mg интрамускулно през ден, общо 10-15 инжекции;
- АТФ - 1 мл интрамускулно през ден, 10-15 инжекции;
- витамин А - 1000 IU/година/ден в 1 доза за 2 седмици;
- Витамин Е - 1 мг/кг/ден в 1 доза в продължение на 2 седмици.
Този вид терапия спомага за подобряване на общото състояние на пациентите, намаляване на тубулните дисфункции и се провежда на курсове 3 пъти годишно.
Левамизол може да се използва като имуномодулатор - 2 мг/кг/ден 2-3 пъти седмично с почивки между дозите от 3-4 дни.
Според данни от изследвания, хипербарната оксигенация има положителен ефект върху тежестта на хематурията и бъбречната дисфункция.
Най-ефективният метод за лечение на наследствен нефрит е навременната бъбречна трансплантация. В този случай не се наблюдава рецидив на заболяването в трансплантирания бъбрек; в малък процент от случаите (около 5%), в трансплантирания бъбрек може да се развие нефрит, свързан с антигени към гломерулната базална мембрана.
Перспективно направление е пренаталната диагностика и генно-инженерната терапия. Експериментите с животни показват висока ефективност на пренасяне на нормални гени, отговорни за синтеза на алфа вериги от колаген тип IV, в бъбречната тъкан, след което се наблюдава синтез на нормални колагенови структури.
Прогноза
Прогнозата за наследствен нефрит винаги е сериозна.
Прогностично неблагоприятни критерии за протичане на наследствен нефрит са:
- мъжки пол;
- ранно развитие на хронична бъбречна недостатъчност при членове на семейството;
- протеинурия (повече от 1 g/ден);
- удебеляване на гломерулните базални мембрани според микроскопия;
- акустичен неврит;
- делеция в гена Col4A5.
Прогнозата за доброкачествена фамилна хематурия е по-благоприятна.
Использованная литература