Медицински експерт на статията

Нови публикации
Остра некротизираща енцефаломиопатия на Лий
Последно прегледани: 04.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
Заболяването е споменато за първи път през 1951 г. Към днешна дата са описани повече от 120 случая. Болестта на Лий (OMIM 256000) е генетично хетерогенно заболяване, което може да се унаследи или ядрено (автозомно рецесивно или Х-свързано), или митохондриално (по-рядко срещано).
 [
[ Причини на синдрома на Лий
Заболяването се основава на дефицит на ензими, осигуряващи производството на енергия, главно поради нарушаване на метаболизма на пирувиновата киселина и дефект в електронния транспорт в дихателната верига. Развива се дефицит на пируват дехидрогеназния комплекс (a-E1 субединица), пируват карбоксилазата, комплекс 1 (NAD-коензим Q-редуктаза) и комплекс 4 (цитохром оксидаза) на дихателната верига.
Установено е, че дефектите на пируват карбоксилазата, комплекс 1 (NAD-коензим Q-редуктаза) и комплекс 4 (цитохром оксидаза) на дихателната верига се унаследяват по автозомно-рецесивен начин, дефектите на пируват дехидрогеназния комплекс (a-E1 субединица) се унаследяват по Х-свързан рецесивен начин. При точкови мутации на мтДНК, които засягат 6-та субединица на АТФазата, типично е митохондриалното унаследяване. Най-често се наблюдава мисценс мутация, свързана със заместване на тимин с гуанин или цитозин в позиция 8993 на мтДНК. По-рядко срещана е мутацията в позиция 9176 на мтДНК. Поради факта, че мутацията T8993G е основният дефект при NARP синдрома, са описани семейства с тези две заболявания. При деца е описана и мутация в мтДНК в позиция 8344, която се среща при MERRF синдром.
Предполага се, че в случай на натрупване на мутантна мтДНК в повечето митохондрии, се развива тежък ход на синдрома на Лий. При митохондриалния генезис на това състояние, мутантна мтДНК се открива в 90% от всички митохондрии. Патогенезата е свързана с нарушение на образуването на енергия в клетките и развитие на лактатна ацидоза.
Симптоми на синдрома на Лий
Първите признаци на заболяването дебютират в ранна възраст (1-3 години). Известни са обаче случаи на проявление на заболяването на 2 седмици и на 6-7-годишна възраст. В началото се развиват неспецифични нарушения: забавено психомоторно развитие, намален апетит, епизоди на повръщане, дефицит на телесно тегло. Впоследствие се засилват неврологичните симптоми: мускулна хипотония или дистония с преход към хипертония, пристъпи на миоклонус или тонично-клонични гърчове, тремор на крайниците, хореоатетоза, нарушение на координацията, намалени сухожилни рефлекси, летаргия, сънливост. Церебралната невродегенерация е прогресивна. Симптомите на пирамидална и екстрапирамидна недостатъчност се засилват, актът на преглъщане е нарушен. Често се наблюдават промени в органа на зрението като птоза, офталмоплегия, атрофия на зрителните нерви, по-рядко пигментна дегенерация на ретината. Понякога се развива хипертрофична кардиомиопатия, появяват се епизоди на тахипнея.
Рядко заболяването протича като остра енцефалопатия. По-типично е хронично или подостро протичане, което води до фатален изход няколко години след началото на заболяването. При бързо протичане (няколко седмици) настъпва смърт в резултат на парализа на дихателния център.
Диагностика на синдрома на Лий
Биохимичен кръвен тест разкрива лактатна ацидоза, дължаща се на натрупване на млечна и пирогроздена киселина в кръвта и цереброспиналната течност, както и повишаване на съдържанието на аланин в кръвта. Нивото на кетонни тела също може да бъде повишено. В урината се открива повишено отделяне на органични киселини: млечна, фумарова и др. Нивото на карнитин в кръвта и тъканите често намалява.
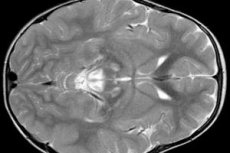
Резултатите от ЕЕГ разкриват фокални признаци на епилептична активност. Данните от ЯМР разкриват уголемяване на мозъчните камери, двустранно мозъчно увреждане, калцификация на базалните ганглии (каудатно ядро, путамен, субстанция нигра, глобус палидус). Може да се открие и атрофия на мозъчните полукълба и мозъчното вещество.
Морфологичното изследване разкрива груби промени в мозъчното вещество: симетрични огнища на некроза, демиелинизация и спонгиозна дегенерация на мозъка, главно на средните отдели, моста, базалните ганглии, таламуса и зрителния нерв. Хистологичната картина включва кистозна дегенерация на мозъчната тъкан, астроцитна глиоза, невронна смърт и увеличаване на броя на митохондриите в клетките. В скелетните мускули се наблюдава натрупване на липидни включвания, намаляване на хистохимичната реакция към комплекси 1 и 4 на дихателната верига, субсарколемално натрупване на митохондрии, анормални митохондрии с дезорганизация на кристите. Феноменът RRF често не се открива.
Как да проучим?
Какви тестове са необходими?
Использованная литература