Медицински експерт на статията

Нови публикации
Ксеродермия пигментозум
Последно прегледани: 04.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
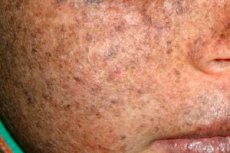
Ксеродерма пигментозум е автозомно-рецесивно генетично заболяване, свързано с възстановяването на ДНК. Заболяването се причинява от мутации в гени, участващи в поправянето на ДНК, увредена от ултравиолетова радиация и други токсични вещества.
Най-често това заболяване засяга деца, които са наричани още „деца на нощта“. Чести усложнения на заболяването са базално-клетъчен карцином и други злокачествени новообразувания на кожата, метастатичен злокачествен меланом и плоскоклетъчен карцином.
 [ 1 ]
[ 1 ]
Причини ксеродерма пигментозум
Патогенеза
Дефицитът на UV ендонуклеазни ензими в клетките на пациента (във фибробластите) или пълното им отсъствие играят важна роля в развитието на заболяването. Този ензим е отговорен за възпроизвеждането на ДНК, увредена от ултравиолетовите лъчи. Според други източници, дефицитът на ензима ДНК полимераза-1 играе важна роля в развитието на заболяването при някои пациенти. Заболяването най-често се развива под въздействието на лъчи с дължина на вълната 280-310 nm. Смята се, че пигментната ксеродерма се развива поради навлизането на различни фотодинамични вещества в тялото или увеличаването на порфирините в човешката биологична среда.
В ранните стадии на заболяването се наблюдават хиперкератоза, атрофия на епидермисното фокусно място, изтъняване на Малпигиевия слой, увеличаване на меланиновите гранули в базалния слой на клетките и хроничен възпалителен инфилтрат в горния слой на дермата (главно около кръвоносните съдове). Впоследствие се откриват дегенеративни промени в колагеновите и еластичните влакна, а на туморен стадий се разкриват признаци, характерни за рак на кожата.
Симптоми ксеродерма пигментозум
Мъжете и жените са еднакво засегнати от това заболяване. Заболяването започва в ранна детска възраст през пролетта или лятото. Кожата на пациента е особено чувствителна към слънчева светлина. Поради това, първият обрив под формата на хиперпигментация, подобна на бенка, се появява върху участъци от кожата, изложени на слънчева светлина. Обривът се увеличава, еритемът се засилва, става тъмнокафяв, появяват се телеангиектазии, ангиоми и атрофия. Впоследствие растат папиломи и брадавици (главно по кожата на лицето и шията), появяват се петна и язви. В резултат на образуването на белези от язви, носът става по-тънък („птичи клюн“), клепачът се извива наопаки. Папиломите обикновено се развиват в злокачествени тумори. Пигментираната ксеродерма, като правило, се среща заедно със спиноцелуларен рак, меланосаркоми.
Какво те притеснява?
Етапи
В клиничния ход на ксеродерма пигментозум се разграничават пет стадия: възпалителен (еритематозен), хиперпигментация, атрофия, хиперкератоза и злокачествени тумори.
По време на възпалителния (еритематозен) стадий, по участъци от кожата, изложени на слънчева светлина (лице, шия, горна част на гърдите, ръце, длани), се появяват подуване, червени петна, а понякога и мехури и везикули. Почти няма обрив по участъци, които не са изложени на слънчева светлина.
При хиперпигментация на мястото на червените петна се появяват светлокафяви, кафяви хиперпигментирани петна, подобни на бенки.
При атрофия кожата изсъхва, изтънява и се набръчква. По кожата на устните и носа се наблюдават множество малки, звездовидни телеангиектазии и белези с лъскава повърхност. Поради атрофията и белезите се развиват микротомия (намаляване на отвора на устата), ектропион, изтъняване на ушите и носа, атрезия на отвора на носа и устата. При 80-85% от пациентите са засегнати очите: отбелязват се конюнктивит, кератит, увреждане на роговицата и лигавицата, отслабване на зрението. По кожата на клепачите се наблюдават дисхромия, телеангиектазия, хиперкератотични промени и тумори.
При хиперкератоза, в описаните по-горе патологични огнища се появяват брадавични тумори, папиломи, кератоакантоми, фиброми, ангиофибриоми и други доброкачествени тумори. Поради това някои учени включват пигментната ксеродерма в групата на предраковите заболявания.
След 10-15 години от началото на заболяването се появяват злокачествени кожни тумори (базалиом, ендотелиом, ангиосарком) в атрофично променени огнища и пигментни петна. Туморите се разрушават за кратко време, метастазират във вътрешни органи и водят до смърт. Във вътрешните органи и тъкани на някои пациенти се наблюдават общи дистрофични промени (синдактелия на втори и трети пръст на крака, зъбна дистрофия, пълна загуба на коса и др.).
Форми
Неврологичната форма на ксеродерма пигментозум се проявява в два синдрома.
Синдромът на Рийд се характеризира с клинични прояви на ксеродерма пигментозум и микроцефалия, идиопатия и бавен растеж на скелетната система. При неврологичната форма на заболяването репарацията на ДНК е трудна и рентгеновата терапия води до увеличаване на основните патологични огнища.
Синдромът на De Sinctis-X Cocchione се характеризира със следните клинични признаци:
- развитие на клинични признаци на ксеродерма пигментозум върху особено фоточувствителна кожа;
- ранна поява на злокачествени тумори;
- едновременно протичане на спастична парализа, микроцефалия и деменция;
- вродени деформации и нанизъм;
- недоразвитие на половите жлези;
- чести спонтанни аборти;
- рецесивно предаване по наследство.
Според някои дерматолози, синдромът на Сантис-Какионе не е самостоятелно заболяване, а тежка и напълно изразена клинична проява на ксеродерма пигментозум.
Генетични форми
Видове |
Джийн |
Локус |
Описание |
Тип A, I, XPA |
XPA |
9q22.3 |
Ксеродерма пигментозум (XP) група А – класическа форма |
Тип B, II, XPB |
XPB |
2q21 |
XP Група Б |
Тип C, III, XPC |
XPC |
3п25 |
XP Група C |
Тип D, IV, XPD |
XPD ERCC6 |
19q13.2-q13.3, 10q11 |
XP група D или синдром на De Sanctis-Cacchione |
Тип E, V, XPE |
ДДБ2 |
11p12-p11 |
XP Група E |
Тип F, VI, XPF |
ERCC4 |
16p13.3-p13.13 |
XP Група F |
Тип G, VII, XPG |
RAD2ERCC5 |
13q33 |
XP група G и COFS синдром (церебро-окуло-фациоскелетен синдром) тип 3 |
Тип V, XPV |
ПОЛХ |
6p21.1-p12 |
Вариант на ксеродерма пигментоза |
Какво трябва да проучим?
Как да проучим?
Към кого да се свържете?
Лечение ксеродерма пигментозум
Антималарийните лекарства (делагил, плаквенил, резоквин и др.), предпазвайки ДНК от ултравиолетовите лъчи, инхибират процеса на деполимеризация, намаляват чувствителността (фотосенсибилизация) на кожата към слънчева светлина. Тези лекарства имат противовъзпалителни и хипосенсибилизиращи свойства. Препоръчва се провеждане на обща терапия заедно с витаминна терапия (B1, B2, PP, B6, B12, A, E), антихистамини (тавегил, дифенхидрамин, супрастин), десенсибилизиращи средства (натриев тиосулфат, 10% калциев хлорид интравенозно 10 ml).
За локално лечение се използват слънцезащитни кремове и мехлеми.
При туморната форма на пигментна ксеродерма се използват хирургични методи, течен азот и лазерни лъчи. За да се предпазите от слънчева светлина, трябва да носите широки дрехи, слънчеви шапки и ръкавици.
Прогноза
Повечето пациенти (2/3) умират преди да навършат 15 години. Някои пациенти могат да живеят до 40-50 години.
[ 20 ]