Медицински експерт на статията

Нови публикации
Субакутна некротизираща енцефаломиопатия Leia
Последно прегледани: 23.04.2024

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
 [
[Причини на синдрома на Leia
Заболяването се основава на дефицит на ензим, придобитата енергия главно от метаболитни нарушения пирогроздена киселина и дефект електронен транспорт в дихателната верига. Пируват дехидрогеназа комплекс дефицит развива (а-субединица Е1), пируват карбоксилаза, комплекс 1 (NAD коензим Q-редуктаза) и комплекс 4 (цитохром оксидаза) дихателната верига.
Установено е, че дефекти пируват, комплекс 1 (NAD коензим Q-редуктаза) и комплекс 4 (цитохром оксидаза) дихателната верига са наследени в автозомно рецесивен начин, дефекти на комплекса пируват дехидрогеназа (а-E1 субединица) - X-свързани рецесивни. Когато мДНК точкови мутации, които засягат 6-АТРаза субединица, митохондриална наследство характеристика. Най-често се среща mistsens мутация, свързана с подмяна на тимин да гуанин или цитозин на позиция 8993 мтДНК. По-рядка е мутация в позиция 9176 mtDNA. Поради факта, че мутации T8993G - основния дефект в синдрома NARP, описан в семейството с наличието на тези две заболявания. Деца също са описани мтДНК мутация в позиция 8344, което се намира в MERRF на синдром.
Предполага се, че в случай на акумулиране на мутантна mtDNA в по-голямата част от митохондриите се развива тежък курс на синдрома на Leia. При митохондриалния генезис на това състояние, мутантната mtDNA се открива в 90% от всички митохондрии. Патогенезата е свързана с нарушаване на производството на енергия в клетките и развитието на лактатна ацидоза.
Симптоми на синдрома на Leia
Първите признаци на дебюта на болестта в ранна възраст (1-3 години). Все пак са известни случаи на проявление на заболяването при 2-седмичните и 6-7-годишните. Първоначално се развиват неспецифични разстройства: забавяне на психомоторното развитие, намален апетит, епизоди на повръщане и липса на телесно тегло. В последващо отглеждане неврологични симптоми: мускулна хипотония или дистония с преминаването към хипертоничност, припадъци, миоклонични гърчове или тонично-клонични припадъци, тремор на крайниците, хореоатетоза, координация разстройство, намалява сухожилни рефлекси, летаргия, сънливост. Церебралната невродегенерация има прогресивен характер. Симптомите на пирамидална и екстрапирамидна недостатъчност се увеличават, преглъщането се нарушава. Често има такава промяна на властта като птоза, офталмоплегия, атрофия на зрителния нерв, пигментен ретинит по-малко. Понякога се развива хипертрофична кардиомиопатия, появяват се епизоди на тахипнея.
Рядко заболяването протича според вида остра енцефалопатия. По-характерно е хроничен или субакутен ток, който води до фатален изход няколко години след началото на заболяването. С бърз поток (няколко седмици), смъртта настъпва в резултат на парализа на дихателния център.
Диагностика на синдрома на Leia
При биохимичен кръвен тест се открива лактатна ацидоза, дължаща се на натрупването на млечни и пирувинови киселини в кръвта и течността, както и повишаване на съдържанието на аланин в кръвта. Също така, нивото на кетонните тела може да се увеличи. В урината има повишена екскреция на органични киселини: млечна, фумарова и др. Нивото на карнитин в кръвта и тъканите често се намалява.
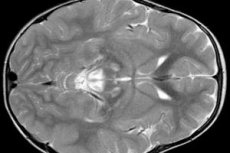
Резултатите от ЕЕГ разкриват фокални признаци на епилептична активност. Според данни от ЯМР се откриват разширяване на вентрикулите на мозъка, двустранно мозъчно увреждане, калцификация на базалните ганглии (ядро на кауда, черупка, черно вещество, бледо топка). Също така е възможно да се идентифицира атрофия на церебралните полукълба и мозъчните вещества.
Морфологични проучване показват големи промени в мозъка вещество: симетричен некроза, демиелинизация и порести дегенерация на мозъка, предимно в средата части, мостове, базалните ганглии, таламуса, оптичен нерв. Хистологичната картина включва циститна дегенерация на мозъчната тъкан, астроцитна глиоза, смърт на неврони, увеличаване на броя на митохондриите в клетките. В скелетната мускулатура - натрупването на липиди включвания намаляване хистохимично реакция на комплекси на 1, 4 дихателната верига на митохондриите конгестия subsarkolemmalnoe, анормални митохондрии с прекъсване на cristae. Явлението RRF често не се открива.
Как да проучим?
Какви тестове са необходими?
Использованная литература