Медицински експерт на статията

Нови публикации
Синдром на Тречър Колинс
Последно прегледани: 04.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.

Вътрематочните нарушения в процесите на развитие на костите причиняват сериозни краниофациални деформации, а една от разновидностите на такава патология е синдромът на Тричър Колинс (TCS) или мандибулофасциалната, т.е. лицево-челюстната дизостоза.
Код на заболяването съгласно МКБ 10: клас XVII (вродени аномалии, деформации и хромозомни нарушения), Q75.4 - мандибулофациална дизостоза.
Причини Синдром на Тречър Колинс
Този синдром е кръстен на изключителния британски офталмолог Едуард Трийчър Колинс, който е описал основните характеристики на патологията преди повече от сто години. Европейските лекари обаче по-често наричат този вид аномалия на лицевите и челюстните кости болест или синдром на Франческети - въз основа на обширните изследвания на швейцарския офталмолог Адолф Франческети, който въвежда термина „мандибулофасциална дизостоза“ в средата на миналия век. В медицинските среди се използва и наименованието синдром на Франческети-Колинс.
Синдромът на Тричър Колинс се причинява от мутации в гена TCOF1 (в локуса 5q31.3-33.3 хромозома), който кодира нуклеоларен фосфопротеин, отговорен за образуването на краниофациалната част на човешкия ембрион. В резултат на преждевременно намаляване на количеството на този протеин, биогенезата и функциите на рРНК се нарушават. Според генетици от изследователската програма „Човешки геном“, тези процеси водят до намаляване на пролиферацията на ембрионални клетки на невралния гребен - гребен по протежение на невралния жлеб, който се затваря в неврална тръба по време на ембрионалното развитие.
Образуването на лицевите тъкани се дължи на трансформация и диференциация на клетки от горната (главната) част на невралния гребен, които мигрират по невралната тръба към областта на първата и втората бранхиална дъга на ембриона. А дефицитът на тези клетки причинява краниофациални деформации. Критичният период за появата на аномалии е от 18 до 28 дни след оплождането. След завършване на миграцията на клетките на невралния гребен (в четвъртата гестационна седмица) се образуват почти всички рехави мезенхимни тъкани в лицевата област, които по-късно (от 5 до 8 седмици) се диференцират в скелетни и съединителни тъкани на всички части на лицето, шията, ларинкса, ухото (включително вътрешното ухо) и бъдещите зъби.
Патогенеза
Патогенезата на синдрома на Тричър Колинс често е фамилна, а аномалията се унаследява по автозомно доминантен начин, въпреки че има случаи на автозомно рецесивно предаване на дефекта (с мутации в други гени, по-специално POLR1C и POLR1D). Най-непредсказуемото нещо при лицево-челюстната дизостоза е, че мутацията се унаследява от деца само в 40-48% от случаите. Тоест, при 52-60% от пациентите причините за синдрома на Тричър Колинс не са свързани с наличието на аномалия в семейството и се смята, че патологията възниква в резултат на спорадични генни мутации de novo. Най-вероятно новите мутации са последствия от тератогенни ефекти върху плода по време на бременност.
Сред тератогенните причини за този синдром, експертите посочват големи дози етанол (етилов алкохол), радиация, цигарен дим, цитомегавирус и токсоплазма, както и хербициди на основата на глифозат (Roundal, Glyfor, Tornado и др.). А списъкът с ятрогенни фактори включва лекарства за акне и себорея с 13-цис-ретиноева киселина (Isotretinoin, Accutane); антиконвулсивното лекарство Фенитоин (Dilantin, Epanutin); психотропни лекарства Диазепам, Валиум, Реланиум, Седуксен.
Симптоми Синдром на Тречър Колинс
В по-голямата си част клиничните признаци на мандибулофасциална дизостоза и степента на тяхното изразяване зависят от характеристиките на проявата на генни мутации. И първите признаци на тази аномалия в повечето случаи са видими при дете веднага след раждането: лицето със синдром на Тричър Колинс има характерен външен вид. Освен това морфологичните аномалии обикновено са двустранни и симетрични.
Най-очевидните симптоми на синдрома на Тричър Колинс са:
- недоразвитие (хипоплазия) на лицевите кости на черепа: зигоматични, зигоматични израстъци на фронталната кост, странични птеригоидни пластинки, параназални синуси, долна челюст и издатини на костните епифизи (кондили);
- недоразвитие на костите на долната челюст (микрогнатия) и мандибуларен ъгъл, който е по-тъп от обичайното;
- носът е с нормален размер, но изглежда голям поради хипоплазия на надбръбните дъги и недоразвитие или липса на зигоматичните дъги в темпоралната област;
- очните процепи са насочени надолу, тоест формата на очите е необичайна, като външните ъгли са увиснали надолу;
- дефекти на долните клепачи (колобом) и частична липса на мигли върху тях;
- неправилно оформени ушни миди с широк диапазон от отклонения, включително разположението им в ъгъла на долната челюст, липса на лобове, слепи фистули между трагуса на ухото и ъгъла на устата и др.;
- стесняване или затваряне (атрезия) на външния слухов канал и аномалии на слуховите костички на средното ухо;
- отсъствие или хипоплазия на паротидните слюнчени жлези;
- фарингеална хипоплазия (стесняване на фаринкса и дихателните пътища);
- несрастване на твърдото небце (цепнатина на небцето), както и отсъствие, скъсяване или неподвижност на мекото небце.
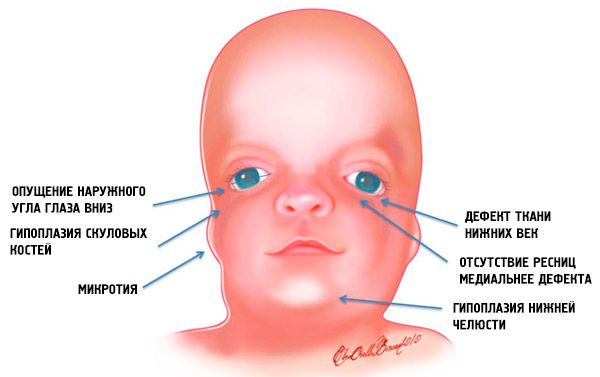
Такива анатомични аномалии във всички случаи имат усложнения. Това са функционални нарушения на слуха под формата на проводима загуба на слуха или пълна глухота; зрителни нарушения поради неправилно формиране на очните ябълки; дефекти на небцето причиняват затруднения с храненето и преглъщането. Съществуват нарушения на зъбната оклузия (малоклузия), свързани с дефекти на челюстта, което от своя страна причинява проблеми с дъвченето и артикулацията. Патологиите на мекото небце обясняват носовия глас.
Усложнения и последствия
Последиците от лицево-челюстните аномалии при синдрома на Тричър Колинс са, че при раждането интелектуалните способности на детето са нормални, но поради слухови дефекти и други нарушения се наблюдава вторична умствена изостаналост.
Освен това, децата с такива дефекти остро усещат своята малоценност и страдат, което се отразява негативно на нервната им система и психиката.
Диагностика Синдром на Тречър Колинс
Постнаталната диагноза на синдрома на Тричър Колинс се основава основно на клинични признаци. Краниофациалната дизостоза се идентифицира лесно, когато синдромът е напълно изразен, но когато са налице минимално изразени симптоми на патология, могат да възникнат проблеми с установяването на правилната диагноза.
В този случай трябва да се обърне специално внимание на оценката на всички функции, свързани с аномалиите, особено тези, засягащи дишането (поради риска от сънна апнея). Ефективността на храненето и кислородната сатурация на хемоглобина също трябва да се оценят и наблюдават.
По-късно, на 5-6-ия ден след раждането, ще е необходимо да се определи степента на увреждане на слуха чрез аудиологично изследване, което трябва да се проведе в родилното отделение.
Предписва се преглед, по време на който се извършва инструментална диагностика чрез флуороскопия на краниофациална дисморфология; пантомография (панорамна рентгенова снимка на костните структури на лицевия череп); пълна черепна компютърна томография в различни проекции; КТ или ЯМР на мозъка за определяне на състоянието на вътрешния слухов канал.
Най-ранната – пренатална – диагностика на лицево-челюстни аномалии при наличие на синдром на Тричър Колинс в семейната анамнеза е възможна чрез биопсия на хорионни въси на 10-11 гестационна седмица (процедурата заплашва спонтанен аборт и инфекция на матката).
Кръвни изследвания се вземат и от членове на семейството; на 16-17 седмица от бременността се анализира околоплодната течност (трансабдоминална амниоцентеза); на 18-20 седмица от бременността се извършва фетоскопия и се взема кръв от феталните съдове на плацентата.
Но най-често ултразвукът се използва при пренатална диагностика на този синдром при плода (на 20-24-та седмица от бременността).
Какви тестове са необходими?
Диференциална диагноза
Същите тези методи се използват от специалистите, когато е необходима диференциална диагностика, за да се разпознае лекият синдром на Тричър Колинс и да се разграничи от други вродени аномалии на краниофациалните кости, по-специално: синдроми на Аперт, Крузон, Нагер, Питърс-Хюелс, Хелерман-Стеф, както и хемифациална микрозомия (синдром на Голдънхар), хипертелоризъм, преждевременно сливане на черепните шевове (краниосиностоза) или нарушено сливане на лицевите кости (краниосиностоза).
Лечение Синдром на Тречър Колинс
Както във всички случаи на генетично обусловени вродени дефекти, лечението на тежките форми на синдрома на Тричър Колинс е изключително палиативно, тъй като просто няма терапевтични методи за такива патологии. Спектърът и степента на деформации при този синдром са обширни и следователно естеството и интензивността на медицинската интервенция също имат много възможности.
Слуховите апарати се използват за коригиране и подобряване на слуха, а сеансите по логопедия се прилагат за подобряване на речта.
Хирургични интервенции се налагат в ранна възраст при тежки случаи на стесняване на дихателните пътища (извършва се трахеостомия) и ларинкса (извършва се гастростомия за хранене). Може да се наложи и хирургична корекция на небцето.
Операциите за удължаване на мандибуларната кост се извършват на възраст 2-3 години или по-късно. Реконструкцията на меките тъкани включва корекция на колобома на долния клепач и аурикуларна пластика.
Прогноза
Каква е прогнозата за тази патология? Тя зависи от степента на деформация и интензивността на симптомите. Синдромът на Тричър Колинс е диагноза, която се поставя през целия живот.
 [ 25 ]
[ 25 ]