Медицински експерт на статията

Нови публикации
Синдром на Мартин-Бел
Последно прегледани: 04.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
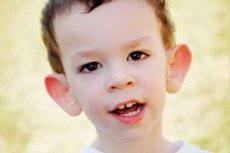
Синдромът на Мартин-Бел е описан през 1943 г. от лекари, на чието име е кръстен. Заболяването е генетично разстройство, състоящо се от умствена изостаналост. През 1969 г. са идентифицирани промени в хромозома X (чупливост в дисталната част на ръката), характерни за това заболяване. През 1991 г. учените откриват гена, отговорен за развитието на това заболяване. Това заболяване се нарича още „синдром на чупливата X“. Както момчетата, така и момичетата са податливи на заболяването, но момчетата са по-често (3 пъти) засегнати.
Епидемиология
Синдромът на Мартин-Бел е доста често срещано заболяване: 0,3-1,0 от 1000 мъже страдат от това заболяване, а 0,2-0,6 от 1000 жени. Освен това, децата със синдром на Мартин-Бел се раждат на всички континенти с еднаква честота. Очевидно е, че националността, цветът на кожата, формата на очите, условията на живот и благосъстоянието на хората не влияят върху появата на заболяването. Честотата му на поява е сравнима само с честотата на синдрома на Даун (1 заболяване от 600-800 новородени). Една пета от мъжете носители на променения ген са здрави, нямат клинични или генни аномалии, останалите имат признаци на умствена изостаналост от леки до тежки форми. Сред жените носители малко повече от една трета са болни.
Синдромът на чупливата X хромозома засяга приблизително 1 на 2500–4000 мъже и 1 на 7000–8000 жени. Разпространението на носителството сред жените се оценява на 1 на 130–250; разпространението на носителството сред мъжете се оценява на 1 на 250–800.
Причини Синдром на Мартин-Бел
Синдромът на Мартин-Бел се развива поради пълното или частично спиране на производството на специфичен протеин от организма. Това се дължи на липсата на отговор от гена FMR1, локализиран в Х хромозомата. Мутацията възниква в резултат на преструктурирането на гена от нестабилни структурни варианти на генните състояния (алели), а не от самото начало. Заболяването се предава само по мъжка линия и мъжът може да не е непременно болен. Мъжките носители предават гена на дъщерите си в непроменен вид, така че умствената им изостаналост не е очевидна. При по-нататъшно предаване на гена от майката на децата ѝ, генът мутира и се появяват всички признаци, характерни за това заболяване.
Патогенеза
Патогенезата на синдрома на Мартин-Бел се основава на мутации в генния апарат, които водят до блокиране на производството на FMR протеин, жизненоважен за организма, особено в невроните, и присъстващ в различни тъкани. Изследванията показват, че FMR протеините участват пряко в процесите на регулиране на транслациите, които се случват в мозъчната тъкан. Липсата на този протеин или ограниченото му производство от организма води до умствена изостаналост.
В патогенезата на заболяването, генната хиперметилация се счита за ключово нарушение, но все още не е възможно окончателно да се определи механизмът на развитие на това разстройство.
Същевременно е открита и локусна хетерогенност на патологията, която е свързана с полиалелизъм, както и полилокус. Установено е наличието на алелни варианти на развитие на заболяването, които са причинени от наличието на точкови мутации, както и от разрушаването на гена от типа FMRL.
Пациентите имат и 2 чупливи триплета, чувствителни към фолиева киселина, разположени на 300 kb, както и 1,5-2 милиона bp от чупливия триплет, съдържащ гена FMR1. Механизмът на мутации, възникващи в гените FRAXE и FRAXF (те са идентифицирани в гореспоменатите чупливи триплети), е свързан с механизма на нарушенията при синдрома на Мартин Бел. Този механизъм се причинява от разпространението на GCC и CGG повторения, които причиняват метилиране на така наречените CpG острови. В допълнение към класическата форма на патологията, съществуват и 2 редки типа, които се различават поради разширяването на тринуклеотидните повторения (при мъжката и женската мейоза).
Установено е, че при класическата форма на синдрома на пациента липсва специален нуклеоцитоплазмен протеин от типа FMR1, който изпълнява функцията на свързване на различни мРНК. Освен това, този протеин насърчава образуването на комплекс, който помага за осъществяване на транслационни процеси вътре в рибозомите.
Симптоми Синдром на Мартин-Бел
Как да разпознаем заболяването при деца? Кои са първите признаци? През първите месеци от живота на детето е невъзможно да се разпознае симптомът на Мартин-Бел, освен че понякога се наблюдава намаляване на мускулния тонус. След една година клиничната картина на заболяването е по-очевидна: детето започва да ходи и говори късно, понякога речта напълно липсва. То е хиперактивно, размахва произволно ръце, страхува се от тълпи и шум, упорит е, има остри изблици на гняв, емоционална нестабилност, появяват се епилептични припадъци, не осъществява зрителен контакт. При пациенти със синдром на Мартин-Бел заболяването се издава и от външния им вид: ушите са стърчащи и големи, челото е тежко, лицето е удължено, брадичката е изпъкнала, страбизъм, широки ръце и крака. Те се характеризират и с ендокринни нарушения: често голямо тегло, затлъстяване, големи тестиси при мъжете, ранен пубертет.

Сред пациентите със синдром на Мартин-Бел нивото на интелигентност варира значително: от лека умствена изостаналост до тежки случаи. Ако нормален човек има коефициент на интелигентност (IQ) средно 100, а гений - 130, то хората, предразположени към заболяването, имат 35-70.
Всички клинични симптоми на патологията могат да бъдат характеризирани с триада от основни прояви:
- олигофрения (коефициент на интелигентност 35-50);
- дисморфофобия (наблюдават се стърчащи уши и прогнатизъм);
- макроорхидизъм, който се появява след настъпването на пубертета.
Приблизително 80% от пациентите имат и пролапс на бикуспидалната клапа.
Пълната форма на синдрома обаче се проявява само при 60% от всички пациенти. При 10% се открива само умствена изостаналост, а при останалите заболяването се развива с различна комбинация от симптоми.
Сред първите признаци на заболяването, които се появяват в ранна възраст:
- болното дете проявява значителна умствена изостаналост в сравнение с развитието на други негови връстници;
- нарушения на вниманието и концентрацията;
- силен инат;
- децата започват да ходят и говорят доста късно;
- наблюдават се хиперактивност и нарушения в развитието на речта;
- много силни и неконтролируеми пристъпи на гняв;
- може да се развие мутизъм - това е пълна липса на реч при дете;
- бебето изпитва социална тревожност и е способно да се паникьосва поради силен шум или други силни звуци;
- детето размахва ръце неконтролируемо и хаотично;
- наблюдава се срамежливост, детето се страхува да не бъде на претъпкани места;
- появата на различни обсесивни идеи, нестабилно емоционално състояние;
- Бебето може да не желае да осъществява зрителен контакт с хората.
При възрастни се наблюдават следните симптоми на патологията:
- специфичен външен вид: удължено лице с тежко чело, големи стърчащи уши, силно изпъкнала брадичка;
- плоскостъпие, отит и страбизъм;
- пубертетът настъпва доста рано;
- може да се развие затлъстяване;
- Доста често се наблюдават сърдечни дефекти при синдрома на Мартин-Бел;
- при мъжете се наблюдава уголемяване на тестисите;
- ставните съединения стават много подвижни;
- теглото и ръстът се увеличават рязко.
Диагностика Синдром на Мартин-Бел
За да диагностицирате синдрома на Мартин Бел, трябва да се свържете с квалифициран генетик. Диагнозата се поставя след специфични генетични тестове, които ви позволяват да идентифицирате дефектната хромозома.
Тестове
В ранен стадий на заболяването се използва цитогенетичен метод, при който от пациента се взема фрагмент от клетъчен материал, към който след това се добавя фолиева киселина, за да се провокират промени в хромозомите. След определен период от време се идентифицира област от хромозомата, където има забележимо изтъняване - това е признак за наличие на синдром на чупливата Х хромозома.
Този тест обаче не е подходящ за диагностициране в по-късните стадии на заболяването, тъй като точността му е намалена от широкото използване на мултивитамини, съдържащи фолиева киселина.
Интегрираната диагностика на синдрома на Мартин-Бел е молекулярно-генетично изследване, което се състои в определяне на броя на така наречените тринуклеотидни повторения в гена.
 [ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
[ 25 ], [ 26 ], [ 27 ], [ 28 ], [ 29 ]
Инструментална диагностика
Високоспецифичен метод за инструментална диагностика е PCR (полимеразна верижна реакция), която позволява да се изследва структурата на аминокиселинните остатъци, съдържащи се в X хромозомата, и по този начин да се определи наличието на синдром на Мартин Бел.
Съществува и отделен, още по-специфичен метод за патологична диагностика – комбинация от PCR и откриване чрез капилярна електрофореза. Този метод е с висока точност и открива хромозомна патология при пациенти с първична яйчникова недостатъчност, както и атаксичен синдром.
Наличието на дефекта може да се установи след извършване на диагностика на ЕЕГ. Пациентите с това заболяване имат подобна биоелектрична мозъчна активност.
Какви тестове са необходими?
Диференциална диагноза
Диференцираните методи, които помагат да се подозира синдромът, включват:
- клинични - 97,5% от пациентите имат явни признаци на умствена изостаналост (умерена или тежка); 62% имат стърчащи големи уши; 68,4% имат голяма изпъкнала брадичка и чело; 68,4% от момчетата имат уголемени тестиси, 41,4% имат речеви особености (неравномерен говор, неконтролируем обем и др.);
- цитогенен - кръвта се изследва за лимфоцитна култура, определя се броят на клетките с чуплива Х хромозома на 100 изследвани клетки;
- Електроенцефалография - записват се промени в електрическите импулси на мозъка, специфични за синдрома на Мартин-Бел.
Към кого да се свържете?
Лечение Синдром на Мартин-Бел
При лечението на възрастни пациенти се използват антидепресанти с психостимуланти. Процесът на лекарствена терапия се наблюдава постоянно от психолог и психиатър. Освен това в частни клиники се извършват микроинжекционни процедури с лекарства като Церебролизин (или негови производни), както и цитомедини (като Солкосерил или Лидаза).
При развитието на атаксичен синдром се използват лекарства за разреждане на кръвта и ноотропици. Освен това се предписват аминокиселинни смеси и ангиопротектори. На жени с първична яйчникова недостатъчност се предписва коригиращо лечение с помощта на билкови лекарства и естрогени.
Антагонисти на глутаминовите рецептори също се използват при лечението.
Традиционно лечението на синдрома на Мартин-Бел включва употребата на лекарства, които повлияват симптомите на заболяването, но не и неговата причина. Тази терапия включва предписване на антидепресанти, невролептици и психостимуланти. Не всички лекарства са показани за употреба при деца, така че списъкът с лекарства е доста ограничен. Невролептиците, които могат да се използват след 3 години (най-ранната възраст за предписването им), включват халоперидол в капки и таблетки, хлорпромазин в разтвор и перициазин в капки. По този начин дозата на халоперидол за деца се изчислява в зависимост от телесното тегло. За възрастни дозата се предписва индивидуално. Приема се перорално, като се започва с 0,5–5 mg 2–3 пъти дневно, след което дозата постепенно се увеличава до 10–15 mg. Когато настъпи подобрение, се преминава към по-ниска доза, за да се поддържа постигнатото състояние. При психомоторно възбуда се предписват 5-10 mg интрамускулно или интравенозно, като са възможни няколко повторения след 30-40 минути. Дневната доза не трябва да надвишава 100 mg. Възможни са странични ефекти под формата на гадене, повръщане, мускулни спазми, повишено кръвно налягане, аритмия и др. Възрастните хора трябва да вземат специални предпазни мерки, тъй като са регистрирани случаи на внезапен сърдечен арест, а също така може да се появи тардивна дискинезия (неволеви движения).
Антидепресантите повишават активността на мозъчните структури, облекчават депресията, напрежението и подобряват настроението. Тези лекарства, препоръчвани за употреба от 5-8-годишна възраст при синдром на Мартин-Бел, включват кломипромин, сертралин, флуоксетин и флувоксамин. По този начин, флуоксетинът се приема перорално по време на хранене 1-2 пъти (за предпочитане през първата половина на деня), като се започва с 20 mg на ден, като се увеличава до 80 mg, ако е необходимо. На възрастните хора не се препоръчва доза по-висока от 60 mg. Курсът на лечение се определя от лекар, но не повече от 5 седмици.
Възможни нежелани реакции: замаяност, тревожност, шум в ушите, загуба на апетит, тахикардия, оток и др. Необходимо е повишено внимание при предписване на пациенти в напреднала възраст, хора със сърдечно-съдови заболявания и диабет.
Психостимулантите са психотропни лекарства, използвани за подобряване на възприятието на външни стимули: те изострят слуха, реакциите и зрението.
Диазепам се предписва като успокоително средство при неврози, тревожност, епилептични припадъци и конвулсии. Приема се перорално, интравенозно, интрамускулно, ректално (в ректума). Предписва се индивидуално, в зависимост от тежестта на заболяването, с най-малки дози от 5-10 mg, дневно - 5-20 mg. Продължителността на лечението е 2-3 месеца. За деца дозата се изчислява, като се вземат предвид телесното тегло и индивидуалните характеристики. Страничните ефекти включват летаргия, апатия, сънливост, гадене, запек. Опасно е да се комбинира с алкохол, възможно е пристрастяване към лекарството.
При лечението на синдрома на Мартин-Бел са наблюдавани случаи на подобрение на състоянието и с въвеждането на лекарства, произведени от животински материал (мозък): церебролизат, церебролизин, церебролизат-М. Основните компоненти на тези лекарства са пептиди, които насърчават производството на протеин в невроните, като по този начин попълват липсващия протеин. Церебролизин се прилага струйно от 5-10 ml, курсът на лечение се състои от 20-30 инжекции. Лекарството се предписва на деца от едногодишна възраст, като се прилага интрамускулно всеки ден по 1-2 ml в продължение на един месец. Възможни са повторни сесии на приложение. Странични ефекти под формата на треска, противопоказано при бременни жени.
Имаше опити за лечение на заболяването с фолиева киселина, но се подобри само поведенческият аспект (нивото на агресия и хиперактивност намаля, говорът се подобри), а на интелектуално ниво нищо не се промени. За подобряване на състоянието на заболяването се предписва фолиева киселина, показани са физиотерапевтични методи, логопедия, педагогическа и социална корекция.
Литиевите препарати също се считат за ефективни, тъй като спомагат за подобряване на адаптацията на пациента към социалната среда, както и на когнитивната дейност. Освен това, те регулират и поведението му в обществото.
Употребата на билки при синдром на Мартин-Бел е възможна като антидепресанти. Билките, които помагат за облекчаване на напрежението, тревожността и подобряване на съня, включват валериана, мента, мащерка, жълт кантарион и лайка. Запарките се приготвят по следния начин: за 1 чаена лъжичка сухи билки ще ви е необходима чаша вряла вода, отварите се запарват поне 20 минути, приемат се предимно вечер преди лягане или следобед. Лъжица мед би била добро допълнение към тях.
Физиотерапевтично лечение
За елиминиране на неврологичните прояви се провеждат специални физиотерапевтични процедури - като упражнения в басейна, мускулна релаксация и акупунктура.
Хирургично лечение
Важен етап от лечението се считат и методите на пластичната хирургия - операции, които спомагат за подобряване на външния вид на пациента. Извършва се пластична хирургия на крайниците и ушните миди, както и на гениталиите. Извършва се и корекция на гинекомастия с еписпадия, както и на други дефекти във външния вид.
Предотвратяване
Единственият метод за предотвратяване на заболяването е пренаталният скрининг на бременни жени. Съществуват специални прегледи, които позволяват ранно откриване на патологията, след което се препоръчва прекъсване на бременността. Като алтернатива се използва ин витро оплождане (IVF), което може да помогне на детето да наследи здрава Х хромозома.
Превенцията на пациента зависи от това дали генната мутация е възникнала отново или е наследена. За тази цел се извършва молекулярно-генетична диагностика. Фактът, че тестът не е разкрил „крехката Х хромозома“ при роднини, говори в полза на „свежестта“ на мутацията, което означава, че рискът от раждане на дете със синдром на Мартин-Бел е много малък. В семейства, където има болни хора, тестът ще помогне да се избегнат повторни случаи.
Прогноза
Прогнозата за синдрома на Мартин-Бел е благоприятна за цял живот, но не и за възстановяване. Продължителността на живота зависи от тежестта на заболяването и свързаните с него дефекти. Пациентът може да живее нормален живот. При тежки форми на синдрома на Мартин-Бел пациентите са изложени на риск от доживотна инвалидност.
Продължителност на живота
Синдромът на Мартин Бел няма сериозно отрицателно въздействие върху здравето, така че продължителността на живота на повечето хора, на които е поставена диагноза тази патология, не се различава от стандартните показатели.