Медицински експерт на статията

Нови публикации
Синдром на Ангелман при деца и възрастни
Последно прегледани: 04.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.

Съществуват редица заболявания, за които изрази като „грижи се за себе си и няма да се разболееш“ звучат, най-малкото, нелепо. Това са патологии, при които някои психически и физически аномалии са присъщи на тялото на детето още преди раждането, но родителите не са виновни за това. Такива заболявания се причиняват от мутации или аномалии в хромозомните набори и се наричат хромозомни или генетични. Синдром на Ангелман, синдром на Даун, синдром на Патау, синдром на Едуардс, синдром на Търнър, синдром на Прадер-Уили - това е само част от генетичните заболявания от доста приличен списък.
Синдром на щастливия човек
Този път ще говорим за патологията, кръстена на английския педиатър Хари Ангелман, който за първи път повдига въпроса за този проблем през 1965 г., след като се сблъсква в практиката си с три необичайни деца, обединени от общи специфични симптоми. Лекарят нарича тези деца деца-кукли и написва статия за тях, която първоначално е озаглавена „Деца-марионетки“. Самата статия и заглавието ѝ са написани под впечатление от картина, видяна в един от музеите на Верона. Картината изобразява смеещо се момче и се казва „Момчето-кукла“. Асоциацията на детето, изобразено на картината, с трите деца, с които Ангелман някога се е сблъсквал в практиката си, подтиква педиатъра да обедини децата в една група поради заболяването, което са имали.
Няма нищо изненадващо във факта, че децата, споменати в статията, не са били забелязани от други лекари. В края на краищата, на пръв поглед изглеждаше, че имат напълно различни заболявания, толкова различна беше общата клинична картина на заболяването в 3 различни случая. Може би „новата“ хромозомна патология би заинтересувала други учени, но по това време генетиката все още не беше достатъчно развита, за да потвърди хипотезата на английския лекар. Следователно, след известен интерес към нея, статията беше хвърлена на задния рафт за дълго време.
Следващото споменаване на синдрома на Ангелман, както сега се наричаше статията на английския педиатър Г. Ангелман, датира от началото на 80-те години на 20-ти век. И едва през 1987 г. беше възможно да се открие причината, поради която малка част от децата се раждат с такива отклонения, че отвън изглеждат постоянно усмихнати и щастливи. Всъщност това изобщо не е вярно и усмивката е просто гримаса, зад която се крие нещастна човешка душа и болката на родителите.
Епидемиология
Според статистиката, хромозомна мутация при дете може да се развие както на фона на подобни мутации при родителите, така и при липса на такива. Няма ясна наследствена природа на синдрома на Ангелман (AS), но вероятността от развитие на патология при родители с хромозомни мутации е доста висока.
Интересно е също, че ако едно семейство вече има дете със синдром на Аспергер, има един процент вероятност да има второ дете със същото разстройство, дори ако родителите са здрави.
Все още няма точна статистика за броя на пациентите със синдром на Ангелман. Може би причината е разнообразието от симптоми, които могат да се проявят в определен състав или изобщо да не се проявят за дълго време. Предполага се, че разпространението на заболяването е: 1 дете на 20 000 новородени. Но тази цифра е много приблизителна.
Причини Синдром на Анжелман
Синдромът на Ангелман е медицинско наименование за хромозомна патология, но далеч не е единственото. Хората наричат това заболяване синдром на куклените деца, синдром на щастливата кукла, синдром на Петрушка и синдром на смеещата се кукла. Измислят се всякакви имена (понякога дори обидни за самите пациенти и техните родители), но болестта си е болест, независимо колко смешно може да изглежда и каквито и да са причините.
И причините за развитието на синдрома на Ангелман, както и много други генетични патологии, във всички случаи са нарушения в структурата на една от хромозомите или на хромозомния набор като цяло. Но в нашия случай целият проблем се крие в хромозома 15, предадена от майката. Тоест, бащината хромозома в този случай няма отклонения, но женската претърпява определени мутации.
Според вида на хромозомната аномалия, синдромът на Ангелман се класифицира като хромозомна мутация. Такива мутации се считат за:
- Делеция (липса на участък от хромозома, съдържащ определен набор от гени; ако един от гените липсва, говорим за микроделеция), която е резултат от две прекъсвания и едно повторно събиране, когато се губи участък от оригиналната хромозома.
- Дупликация (наличието на допълнителен участък в хромозома, който е копие на съществуващ), която в повечето случаи води до смърт на човек, а по-рядко до безплодие.
- Инверсия (обръщане на един от участъците на хромозомата на 180 градуса, т.е. в обратна посока, като след това гените в нея се разполагат в обратен ред), когато прекъснатите краища на хромозомата се свързват в ред, различен от първоначалния.
- Вмъкване (ако част от генетичния материал в хромозомата е не на мястото си),
- транслокация (ако определен участък от хромозома е прикрепен към друг хромозом; такава мутация може да бъде взаимна без загуба на участъци).
Получавайки мутирала хромозома от нищо неподозираща майка, детето е обречено да се роди с аномалии. Най-честата причина за синдрома на Ангелман все още се счита за делеция на майчината 15-та хромозома, когато липсва малка секция. По-рядко срещани мутации при синдрома на „смеещата се кукла“ се считат за:
- транслокация,
- еднобащинска дизомия (ако детето е получило чифт хромозоми от бащата, майчината хромозома липсва),
- мутация на гени в ДНК, които са едновременно основен градивен (генетичен) материал и инструкции за правилното му използване (по-специално, мутация на гена ube3a в майчината хромозома).
Наличието на една от тези мутации при родителите е рисков фактор за развитието на синдрома на Ангелман при деца. Но не само хромозомните мутации, но и геномните (които са свързани с количествена промяна в хромозомните набори и са по-чести от хромозомните) могат да провокират развитието на заболяването при дете. Често срещаните геномни мутации включват хромозомна тризомия (ако хромозомният набор на човек има повече от 46 хромозоми).
За да се появи патология при дете, изобщо не е необходимо родителите да имат хромозомни аномалии. И все пак, има определен процент пациенти, чието заболяване е наследствено.
Патогенеза
Нека се задълбочим малко в биологията, или по-точно в генетиката. Генетичната информация на всеки отделен човешки организъм се съдържа в 23 двойки хромозоми. Едната хромозома от двойката се предава на детето от бащата, другата - от майката. Всички двойки хромозоми се различават по форма и размер и носят определена информация. Така 23-тата двойка хромозоми (X и Y хромозоми) е отговорна за формирането на половите характеристики на бебето (XX - момиче, XY - момче, докато Y хромозомата може да бъде получена от детето само от бащата).
В идеалния случай детето получава от родителите си 46 хромозоми, които формират неговите генетични характеристики, предопределяйки го като индивид. По-голям брой хромозоми се нарича тризомия и се счита за отклонение от нормата. Например, наличието на хромозома 47 в хромозомния набор (кариотип, определящ вида и индивидуалните характеристики) причинява появата на синдром на Даун.
Ако хромозомите се оцветят със специално багрило, тогава под микроскоп можете да видите ивици с различни нюанси по всяка от тях. Вътре във всяка ивица има огромен брой гени. Всички тези ивици са номерирани от учените и имат фиксирано местоположение. Липсата на една от ивиците се счита за отклонение от нормата. При синдрома на Ангелман много често може да се наблюдава липсата на сегменти от майчината хромозома в интервала q11-q13, разположени в дългото рамо, броят на ДНК базите в които е само около 4 милиона.
Основният компонент на хромозомата се счита за невероятно дълга ДНК молекула, съдържаща хиляди гени и десетки и стотици милиони азотни бази. Така хромозома 15, отговорна за развитието на синдрома на Ангелман и няколко други, съдържа 1200 гена и около 100 милиона бази. Всякакви нарушения в структурата на ДНК молекулата със сигурност ще повлияят на външния вид и развитието на бъдещото дете.
Генетичната информация, съдържаща се в гените, се преобразува в протеин или РНК. Този процес се нарича генна експресия. По този начин генетичната информация, получена от родителите, получава едновременно форма и съдържание, което се въплъщава в техния уникален женски или мъжки наследник.
Съществуват редица патологии с некласически тип наследяване, включително синдром на Ангелман, при който гените, получени от родителите като част от сдвоени хромозоми, носят уникален отпечатък на родителите и се проявяват по различни начини.
И така, синдромът на Ангелман е ярък пример за геномно импринтинг, при който генната експресия в тялото на детето е пряко зависима от това от кой родител са получени алелите (различни форми на един и същ ген, получени от бащата и майката, разположени върху идентични участъци от сдвоени хромозоми). Тоест, само аномалии в майчината хромозома водят до развитието на синдрома, докато мутациите и структурните нарушения на бащината хромозома причиняват напълно различни патологии.
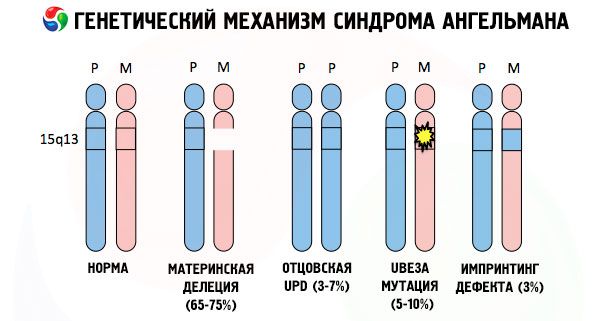
При тази патология се наблюдава липса на определени гени в майчината хромозома или загуба/намаляване на активността на отделни гени (в по-голямата част от случаите, генът ube3a, който участва в метаболизма на убиквитин, протеин, регулиращ разграждането на други протеини). В резултат на това детето се диагностицира с аномалии в психичното развитие и физически деформации.
Симптоми Синдром на Анжелман
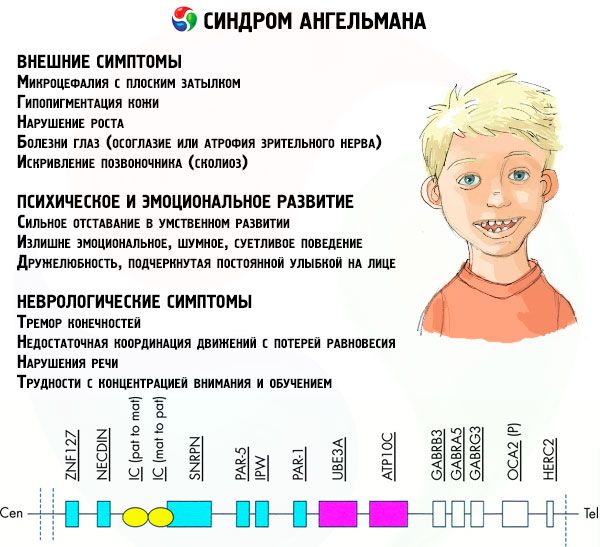
Симптомите на синдрома на Ангелман засягат различни аспекти от живота и развитието на детето: физически, неврологични, психически. Въз основа на това могат да се идентифицират 3 групи симптоми, които показват развитието на тази патология.
- Външни или физически симптоми:
- непропорционално малка глава в сравнение с тялото и крайниците, които са с нормален размер,
- твърде широка уста,
- почти винаги има усмивка на лицето (с отворена уста),
- редки зъби,
- тясна горна устна,
- често изпъкващ широк език,
- изпъкнала долна челюст,
- заострена брадичка,
- много светла кожа, често окосмяване (албинизъм, свързан с факта, че тялото не произвежда пигмента меланин),
- тъмни петна по светла кожа (хипопигментация поради недостатъчно производство на меланин)
- физически или външни симптоми: очни заболявания като страбизъм или атрофия на зрителния нерв,
- изкривяване на гръбначния стълб (сколиоза),
- сковани крака (при ходене човек не сгъва краката си в коленете поради ниска подвижност на ставите, откъдето идва и сравнението с походката на кукла).
- Симптоми, свързани с психическото и емоционалното развитие:
- тежка умствена изостаналост,
- прекалено емоционално, шумно, суетливо поведение,
- често пляскане с ръце,
- изразена дружелюбност, подчертана от постоянна усмивка на лицето,
- чест смях без причина.
- Неврологични симптоми:
- тремор на крайниците,
- недостатъчна координация на движенията със загуба на равновесие,
- намален мускулен тонус,
- различни нарушения на съня,
- чести истерични пристъпи в детството,
- нарушения на речта (детето започва да говори късно, има лоши комуникативни умения и неясна реч),
- хиперактивност на фона на повишена възбудимост,
- трудности с концентрацията и ученето.
Но това е обобщена картина на заболяването. Всъщност клиничната картина на синдрома на Ангелман до голяма степен зависи от стадия на развитие на заболяването и вида на хромозомната мутация, която е причинила патологията. Това означава, че симптомите на заболяването могат да се различават значително при различните пациенти, което дълго време не ни позволяваше да различим патологията от други с подобна клинична картина.
Сред общия брой симптоми можем да подчертаем тези, които са характерни за всички пациенти без изключение:
- тежка умствена изостаналост,
- неподходящо поведение (неразумен смях, повишена възбудимост, лоша концентрация, състояние на еуфория),
- недоразвитие на двигателните умения,
- лоша координация на движенията, атаксия на походката (неравномерно темпо, люлеене от едната страна на другата и др.), тремор на крайниците.
- нарушение на речевото развитие с преобладаване на невербални средства за комуникация.
Сред симптомите, срещани при по-голямата част от пациентите, могат да се разграничат следните:
- диспропорция между главата и тялото, причинена от забавено физическо развитие,
- при много пациенти формата на черепа е такава, че размерът на мозъка остава по-малък, отколкото при здрави хора (микроцефалия),
- епилептични припадъци преди 3-годишна възраст с прогресивно намаляване на силата и честотата в по-напреднала възраст,
- изкривяване на ЕЕГ параметрите (колебания и висока амплитуда на нискочестотни вълни).
Тези симптоми са доста често срещани, но 20% от пациентите със синдром на Ангелман не ги имат.
Още по-рядко е възможно да се диагностицират такива прояви на заболяването като:
- тежък или лек страбизъм,
- лош контрол върху движението на езика, което води до това пациентите често да изпъват езика си без причина,
- затруднения с преглъщането и сукането, особено при малки деца,
- нарушаване на пигментацията на кожата и очите,
- ръце, повдигнати или свити по време на ходене,
- хиперрефлексия,
- нарушения на съня, особено в детска възраст,
- често слюноотделяне,
- неутолима жажда,
- прекалено активни дъвкателни движения,
- свръхчувствителност към топлина,
- плосък тилен край на главата,
- изпъкнала долна челюст,
- гладки длани.
Доста голям процент от пациентите имат проблеми с уринирането, което слабо контролират, нарушена фина моторика, което създава трудности при самообслужване и учене, и наднормено тегло. Почти всички пациенти преживяват пубертета по-късно от здравите си връстници.
Децата със синдром на Ангелман възприемат добре устната реч и я разбират, но не желаят да участват в разговор, ограничавайки речта си до няколко десетки думи, необходими в ежедневието. В зряла възраст обаче такива пациенти изглеждат по-млади от връстниците си без генетични патологии.
Много от симптомите на синдрома на Ангелман са непостоянни, така че клиничната картина на заболяването се променя значително с възрастта. Конвулсиите и епилептичните припадъци стават по-редки или изчезват напълно, пациентът става по-малко възбудим, а сънят се подобрява.
Усложнения и последствия
Синдромът на Ангелман е тежка, в момента практически нелечима хромозомна патология, която лишава пациентите от възможността да живеят нормален живот. Какъв ще бъде животът на дете със синдром на Ангелман, до голяма степен зависи от вида на хромозомната аномалия.
Дублирането на хромозомен сегмент е несъвместимо с живота в повечето случаи. И дори ако такива пациенти не умрат в ранна детска възраст и достигнат пубертет, те нямат шанс да имат деца.
Заличаването или отсъствието на част от гените, което се среща най-често при синдрома на Ангелман, е пречка за детето да се научи да ходи и говори. Такива деца имат по-тежка форма на умствена изостаналост, а епилептичните припадъци се появяват по-често и интензивността им е много по-голяма, отколкото при пациенти с други хромозомни аномалии.
Ако има само мутация на един ген, с нужното внимание и подход детето може да бъде преподано на основите на самообслужването, комуникацията и взаимодействието в група, въпреки че все още ще изостава от връстниците си в развитието.
За децата със синдром на Ангелман, които са добри по природа, най-важното е любовта и вниманието на родителите им. Само в този случай образованието на детето ще даде плодове, дори и малки. Разбира се, пациентите със синдром на Ангелман няма да могат да учат в редовно училище. Те се нуждаят от специални класове, където децата първо ще бъдат научени да се концентрират, а след това постепенно ще им бъдат дадени основите на училищните знания.
Диагностика Синдром на Анжелман
Синдромът на Ангелман е вродена патология на развитието. Но поради определени обстоятелства често е невъзможно да се диагностицира в ранна детска възраст. Това се дължи на неспецифичността и слабата изява на симптомите при кърмачета и деца под 3-годишна възраст. А разпространението на заболяването в нашата страна не е толкова голямо, че лекарите да са се научили да го разпознават сред неговите връстници.
Синдромът на Ангелман при кърмачета може да се прояви като намален мускулен тонус, което се проявява в проблеми с храненето (слабост на сукателния и преглъщащ рефлекс), а по-късно и в трудности при ходене (такива деца започват да ходят много по-късно). Тези симптоми са първите признаци на аномалия в развитието на бебето, която може да е свързана с хромозомна аномалия. Само генетичен анализ може да потвърди това предположение.
Специално внимание се обръща на деца, чиито родители имат различни геномни или хромозомни нарушения. В крайна сметка, заболяването може да не се прояви в началото и ако патологията се открие навреме, като се започне интензивна работа с детето, е възможно да се постигне значително по-голям успех в обучението, забавяйки прогресията на заболяването.
Ако родителите имат различни хромозомни аномалии, генетичният анализ се извършва още преди раждането на бебето, тъй като SA е една от патологиите, които могат да бъдат открити в ембрионалния стадий.
Събирането на материал за генетични изследвания може да се извърши по два начина:
- инвазивно (с определен процент риск, тъй като е необходимо проникване в матката, за да се вземе проба от околоплодна течност),
- неинвазивен (анализ на ДНК-то на бебето от кръвта на майката).
След това се провеждат следните изследвания:
- флуоресцентна in situ хибридизация (FISH метод) – свързване на ДНК сонда, маркирана със специално багрило, към изследваната ДНК, последвано от изследване под микроскоп.
- анализ на мутации в гена ube3a и импринтирани гени,
- Анализ на ДНК метилирането с помощта на специални методи, използвани в генетиката.
Генетичните тестове предоставят сравнително точна информация в случай на хромозомни аномалии, което означава, че бъдещите родители знаят предварително за какво да се подготвят. Има обаче изключения. При определена група пациенти, при наличие на всички симптоми, показващи патология, резултатите от теста остават нормални. Тоест, патологията може да бъде идентифицирана само чрез внимателно наблюдение на детето от ранна детска възраст: как се храни, кога е започнало да ходи и говори, дали сгъва краката си при ходене и т.н.
В допълнение към метода FISH, сред инструменталните диагностични методи за синдрома на Ангелман, може да се разграничат томография (КТ или ЯМР), която помага да се определи състоянието и размера на мозъка, и електроенцефалограма (ЕЕГ), която показва как работят отделните части на мозъка.
Лекарите обикновено поставят окончателна диагноза на възраст 3-7 години, когато пациентът вече има повечето симптоми и динамиката на развитие на заболяването е видима.
Какви тестове са необходими?
Диференциална диагноза
Синдромът на Ангелман е генетична патология, която практически няма специфични прояви. Повечето симптоми могат еднакво да показват както AS, така и други генетични патологии.
Диференциалната диагноза на синдрома на Ангелман се провежда със следните патологии:
- Синдром на Пит-Хопкинс (пациентите се характеризират с умствена изостаналост, весел характер, усмивки, имат доста голяма и широка уста, отбелязва се микроцефалия). Разликата е в пристъпи на хипервентилация и задържане на дишането в будно състояние.
- Синдром на Кристиансън (пациентите са умствено изостанали хора с весел нрав, неспособни да говорят, характеризиращи се с микроцефалия, атаксия, конвулсии, неволеви мускулни движения).
- Синдром на Моват-Уилсън (симптоми: умствена изостаналост, епилептични припадъци, заострена брадичка, отворена уста, щастливо изражение на лицето, микроцефалия). Отличителни черти: голямо разстояние между очите, очи, наклонени навътре, заоблен връх на носа, обърната назад ушна мида.
- Синдром на Кабуки (характеризиращ се с лека до умерена умствена изостаналост, говорни и двигателни проблеми, мускулна слабост, епилептични припадъци, микроцефалия, дълги интервали между сърбежите и нарушена координация). Характеризира се с извити вежди, обърната странична част на долния клепач, широко разположени очи, дълги палпебрални фисури с дълги, гъсти мигли.
- Синдром на Рет (диференциация от AS при жени). Симптоми: забавено развитие на речта, гърчове, микроцефалия. Разликата е, че няма щастливо изражение на лицето, има пристъпи на апнея и апраксия, които прогресират с времето.
- Автозомно-рецесивен синдром на умствена тардация 38 (симптоми: изразена умствена изостаналост със забавяне на двигателните умения и речта, мускулна слабост, проблеми с храненето в ранна детска възраст, импулсивност). Отличителна черта е синият цвят на ириса.
- Синдром на дублиране на гена MECP 2 (диференциация от SA при мъжете). Симптоми: тежка умствена изостаналост, мускулна слабост от детството, проблеми с говора или липса на говор, епилепсия. Отличителни черти: прогресивна миопатия, постоянно повтарящи се инфекции.
- Синдром на Клифстра (симптоми: проблеми с говора и мисленето, мускулна слабост, нарушения на съня, липса на внимание, отворена уста, хиперактивност, гърчове, атаксия, нарушения на равновесието). Отличителни черти: плоско лице, къс чип нос, широко разположени очи, голяма извита долна устна, агресивни изблици.
- Синдром на Смит-Магенис (характеризиращ се с гърчове, проблеми със съня, нарушения в интелектуалното и двигателното развитие). Отличителните черти включват широко и плоско лице и изпъкнало чело.
- Синдром на Кулен-де Врис (лека до умерена умствена изостаналост, мускулна слабост, гърчове, дружелюбност). Отличителни черти: дълго лице с високо чело, стърчащи уши, наклонени очи, висока подвижност на ставите, вродени сърдечни дефекти.
- Синдром на Фелан-Макдермид (симптоми: умствена изостаналост, нарушения на говора или липса на говор). Отличителни черти: големи ръце с развити мускули, мускулна слабост от раждането, слабо изпотяване.
Патологии като дефицит на аденил сукцинат, синдром на автозомно-рецесивна умствена изостаналост 1, синдром на дупликация на хромозома 2q23.1, синдроми на хаплонедостатъчност на гените FOXG1, STXBP1 или MEF2C и някои други могат да се „похвалят“ със симптоми, подобни на синдрома на Ангелман.
Задачата на лекаря е да постави точна диагноза, като диференцира синдрома на Ангелман от патологии с подобни симптоми и да предпише ефективно лечение, което е релевантно на диагностицирания стадий на заболяването.
Лечение Синдром на Анжелман
Синдромът на Ангелман е една от онези патологии, за които медицината все още търси ефективно лечение. Етиологичното лечение на заболяването е в етап на разработка с различни методи и средства, много от които все още не са тествани върху хора. Това означава, че засега лекарите трябва да се ограничат до симптоматична терапия, която помага по някакъв начин да облекчи незавидното положение на деца и възрастни със синдром на марионетката, страдащи от епилептични припадъци, слюноотделяне, хипотония и нарушения на съня.
По този начин е възможно да се намали честотата и силата на епилептичните припадъци с помощта на правилно подбран антиконвулсивен медикамент. Но цялата трудност е, че припадъците при пациенти със СА се различават от обикновените епилептични припадъци по това, че се характеризират с няколко вида припадъци, което означава, че състоянието може да се облекчи чрез едновременно приложение на няколко лекарства.
Най-популярните антиконвулсанти, използвани за лечение на синдрома на Ангелман, са: валпроева киселина, топирамат, ламотригин, леветирацетам, клоназепам и лекарства на тяхна основа. По-рядко се използват лекарства на основата на кармазепин, фенитоин, фенобарбитал, етосуксимид, тъй като някои от тях могат да провокират парадоксален ефект, състоящ се в засилване и увеличаване на честотата на епилептичните припадъци. Това се случва, ако лекарството се използва като част от монотерапия.
За лечение на лигавенето обикновено се използват два метода: медикаментозен (лекарства, които потискат производството на слюнка) и хирургичен, който включва реимплантация на слюнчените каналчета. Но в случай на саркома, тези методи се считат за неефективни и въпросът остава открит. Родителите и тези, които се грижат за такива пациенти, трябва да обърнат специално внимание на този проблем, тъй като самите пациенти обикновено не контролират лигавенето, а някои просто не са в състояние да се грижат сами за себе си.
Друг проблем е кратката продължителност на съня. Често децата със синдром на Ангелман спят не повече от 5 часа, което се отразява негативно на функционирането на целия организъм. Лесно възбудимите, активни деца, които обичат игрите и общуването (дори и да се опитват да се ограничат до невербални методи), са забележимо уморени през деня. За да си почине добре, тялото се нуждае от дълбок, пълноценен сън, но точно в това е уловката.
Изглежда, че седативните лекарства (фенотиазини и атипични антипсихотици), които успокояват нервната система, би трябвало да са достатъчни за подобряване на съня при възбудими пациенти. Но в случай на стеноз на Аспергер, употребата на такива лекарства е изпълнена с появата на негативни ефекти. Затова лекарите все още предпочитат леки сънотворни, като например Мелатонин (естествен хормонален препарат на базата на хормона на съня), който се дава на пациентите час преди лягане в количество от 1 таблетка, и Дифенхидрамин. Честотата на приложение и дозировката му се определят от лекаря в зависимост от състоянието и възрастта на пациента.
Понякога пациентите със синдром на Ангелман имат проблеми с храносмилането и изпражненията. Можете да подобрите изпражненията си с лаксативи (за предпочитане билкови).
Или можете да подходите към проблема по различен начин, както направиха американските лекари, въз основа на някои методи за лечение на аутизъм, защото много симптоми, характерни за AS, са характерни и за аутизма (импулсивност, неволеви движения, повтарящи се действия, дефицит на вниманието, проблеми с комуникацията и др.). Беше отбелязано, че въвеждането на хормона секретин, който нормализира храносмилането и изпражненията, има положителен ефект върху вниманието на пациентите, а окситоцинът помага за подобряване на когнитивните способности и паметта на детето и коригиране на поведението.
Вярно е, че само хормоните не са достатъчни, особено когато става въпрос за деца. При синдрома на Ангелман са показани поведенческа терапия, работа с психолог и логопед (преподаване на невербални методи за комуникация и жестомимичен език). Образованието на такива деца трябва да се основава на индивидуална програма с участието на специално обучени учители, психолог и родители. За съжаление, това не е възможно навсякъде и семействата остават сами с проблема си.
Тъй като много млади пациенти с АС страдат от нисък мускулен тонус и проблеми със ставите, се обръща голямо внимание на физиотерапията. Най-често лекарите прибягват до използването на парафинови апликации, електрофореза и магнитна терапия.
Активният тонизиращ масаж и специалните упражнения по лечебна физкултура ще помогнат на болното дете след известно време да се изправи на крака и да ходи уверено. Особено полезна в това отношение е аквагимнастиката, която се препоръчва при СА в хладна вода. Тя повишава мускулния тонус и учи детето да контролира тялото си и да координира движенията.
Антиконвулсивно лечение
Най-опасният симптом на синдрома на Ангелман са припадъци, подобни на тези при епилепсия. Този симптом се наблюдава при 80% от пациентите, което означава, че на всички тях е необходимо да им бъде предписано ефективно антиконвулсивно лечение.
Лечението на епилептични припадъци се извършва с помощта на витамини и антиконвулсанти. При синдром на Ангелман, придружен от конвулсивен синдром, витамини от група В, както и витамини C, D и E ще бъдат полезни. Но самостоятелното предписване на витаминна терапия в този случай е много опасно, тъй като неконтролираният прием на витамини може да намали ефективността на антиепилептичните лекарства и да провокира нови, по-тежки и продължителни припадъци.
Изборът на антиконвулсивни лекарства и предписването на ефективната им доза също трябва да се извършва от лекар специалист. Той или тя решава и дали едно лекарство ще бъде достатъчно или пациентът ще трябва да приема 2 или повече лекарства за продължителен период от време.
За повечето пациенти лекарите предписват лекарства с валпроева киселина (Валпроева киселина, Депакин, Конвулекс, Валпарин и др.), които предотвратяват гърчовете и подобряват настроението и психическото състояние на пациентите.
Валпроевата киселина се предлага под формата на таблетки, сироп и инжекционни разтвори. Най-популярното лекарство е лекарството с удължено освобождаване "Депакин" в таблетки и като разтвор за интравенозно приложение. Дозировката на лекарството се определя от лекаря индивидуално в зависимост от теглото, възрастта и състоянието на пациента.
Лекарството се приема по време на хранене 2 до 3 пъти дневно. Средната дневна доза е 20-30 мг на 1 килограм тегло на пациента, максималната е 50 мг/кг на ден.
Противопоказания за употреба. Да не се използва при нарушена функция на черния дроб и панкреаса, хеморагична диатеза, хепатит, порфирия и свръхчувствителност към лекарството.
Страничните ефекти включват тремор на ръцете, храносмилателни и изпражнетелни нарушения, както и промени в телесното тегло.
"Топирамат" също е лекарство по избор за SA. Произвежда се под формата на таблетки и се използва както като част от монотерапия, така и в комбинация с други лекарства.
Начин на приложение и дозировка. Таблетките се приемат перорално, независимо от приема на храна. Началната дневна доза за възрастни е 25-50 mg, за деца - 0,5-1 mg/kg. Всяка седмица дозата се увеличава според указанията на лекаря.
Лекарството не трябва да се приема по време на бременност и кърмене, както и при свръхчувствителност към неговите компоненти. Лекарството има много различни странични ефекти.
Лекарства, които лекар може да предпише за синдром на Ангелман: Кломазепам, Ривотрил, Ламотригин, Сейзар, Ламиктал, Леветирацетам, Кепра, Епитера и др.
 [ 16 ], [ 17 ], [ 18 ], [ 19 ]
[ 16 ], [ 17 ], [ 18 ], [ 19 ]
Традиционна медицина и хомеопатия
Традиционната медицина, подобно на хомеопатичните препарати, разбира се, е относително безопасна, но ефективността на подобно лечение за синдрома на Ангелман може да се счита за спорна.
Въпреки че народното лечение все пак може да помогне в някои неща. Говорим за спиране на епилептичните припадъци. В това отношение билколечението може да бъде доста ефективно.
Добър ефект се осигурява от лечебна колекция на базата на божур, женско биле и водна леща (компонентите се приемат в равни количества). Билките трябва да се смилат на брашно. След 2 седмици от началото на приема можете да забележите значително намаляване на честотата на гърчовете.
Отвара от лавандула (1 чаена лъжичка на чаша вряла вода) също е полезна при крампи. Сместа се вари 5 минути и се запарва за половин час. Лекарството се приема през нощта в продължение на 14 дни.
Водна (или алкохолна) инфузия от майчина жълта тръстика се счита за ефективна при епилептични припадъци.
От хомеопатичните препарати за предотвратяване на гърчове при синдром на Ангелман можете да използвате лекарства на базата на лайка и дъвка, Acidum hydrocyanicum, Argentum nitricum, Kalium bromatum, Arsenicum album. Но трябва да се има предвид, че само хомеопатичен лекар може да предпише ефективни и безопасни дози лекарства във всеки конкретен случай.
Предотвратяване
Както читателят вероятно вече е разбрал, медицината все още не е в състояние да предотврати генни мутации и други хромозомни аномалии, както и да коригира ситуацията. Това може да се случи на всеки, защото децата със синдром на Ангелман се раждат от здрави родители, а генетиката, която в момента е един от най-слабо изучените клонове на медицината, все още не може да обясни това.
Единственото, което може да се направи, е да се подходи отговорно към планирането на бременността, да се регистрираме и да се подложим на прегледи навреме. Но отново, подобна мярка ще бъде по-скоро образователна, отколкото превантивна, както всеки преглед. Но младите родители ще знаят предварително за какво да се подготвят и в случай на положителен отговор, ще решат дали могат да поемат такава отговорност като отглеждането на болно дете.
Прогноза
Прогнозата за синдрома на Ангелман зависи от естеството на хромозомната аномалия и навременността на нейното откриване. Най-засегнати са децата, чиято хромозома 15 съдържа „празнини“ в гените (делеция). Вероятността такива пациенти да проходят и да говорят е изключително ниска. Други случаи могат да бъдат коригирани с внимателен подход и любов към детето.
За съжаление, такива пациенти няма да могат да станат пълноправни членове на обществото, въпреки факта, че далеч не са глупави, разбират речта и нейното значение. Те обаче ще имат проблеми с комуникацията до края на живота си. Пациентите могат да бъдат обучавани на жестомимичен език от детството, но не могат да бъдат принуждавани да общуват с думи. Речникът на „говорещите“ пациенти е ограничен до минимума от думи, използвани в ежедневието (5-15 думи).
Що се отнася до продължителността на живота и общото здравословно състояние на пациентите със синдром на Ангелман, тук цифрите се колебаят около средните стойности. В зряла възраст пациентите се сблъскват най-вече със здравословни проблеми като сколиоза и затлъстяване, които при правилен подход към лечението не са животозастрашаващи.