Медицински експерт на статията

Нови публикации
Алкаптонурията е вродена ензимна аномалия
Последно прегледани: 04.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
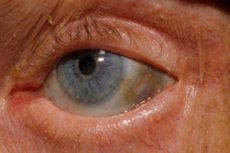
Едно от много редките метаболитни нарушения, алкаптонурия, се отнася до вродени аномалии в метаболизма на аминокиселината тирозин.
Този синдром може да се нарече още дефицит на хомогентизатоксидаза, хомогентизинурия, наследствена охроноза или болест на черната урина.[ 1 ]
Епидемиология
Според статистиката, на 1 милион души има не повече от девет случая на алкаптонурия. А в повечето европейски страни има един случай на 100-250 хиляди живородени деца.
Сред европейските страни изключение прави Словакия (особено относително малкият северозападен регион), където разпространението на алкаптонурия е един случай на 19 000 новородени. Това най-вероятно се дължи на факта, че сред словашките ромски семейства, живеещи там, нивото на инбридинг (бракове между братовчеди) е най-високото в Европа: 10-14%. [ 2 ]
Причини алкаптонурия
Точните причини за алкаптонурията, като вродено нарушение на катаболизма (метаболитния разпад) на ароматната (хомоциклична) α-аминокиселина тирозин, са установени: този тип метаболитно разстройство е следствие от хомозиготни или съставни хетерозиготни мутации на един от хилядите гени на хромозома 3, по-точно на гена HGD в локус 3q21-q23 на дългото рамо на хромозомата. Този ген кодира нуклеотидните последователности на чернодробния ензим хомогентизат-1,2-диоксигеназа [ 3 ] (наричан още хомогентизинова киселинна оксидаза или хомогентизат оксидаза) - желязосъдържащ металопротеин, необходим за един от етапите на разграждане на тирозина в организма. [ 4 ], [ 5 ]
Следователно, алкаптонурията е дефект на ензима хомогентизат-1,2-диоксигеназа, или по-точно, резултат от неговия генетично обусловен дефицит или пълно отсъствие. [ 6 ]
Като вроден ензимен дефицит, алкаптонурията се наследява като автозомно-рецесивен белег, т.е. за да се появи алкаптонурия при деца, и двамата родители трябва да имат модифициран ген за ензима, тъй като всеки от тях предава на детето само едно копие на гена от двете налични.
Според последните данни има повече от двеста варианта на модификация на гена HGD, като най-често се наблюдават миссенс мутации, транслокация и сплайсинг.
Рискови фактори
Единственият рисков фактор за развитие на тази вродена ензимопатия е наличието ѝ в семейната анамнеза и наследяването на две модифицирани копия на HGD гена, ако родителите не проявяват алкаптонурия (рискът от предаване на аномалията е 25%) или единият от родителите има това заболяване. [ 7 ]
Патогенеза
Тирозинът играе ключова роля в синтеза на протеини, производството на хромопротеини – кожния пигмент меланин, както и на тироидни хормони и катехоламинови невротрансмитери.
Механизмът за регулиране на количеството тирозин в клетките е много сложен и тялото нормализира излишното му съдържание чрез разграждането му. Процесът на катаболизъм на тирозина, както при всички ароматни аминокиселини, е многоетапен и протича на няколко етапа. Всеки етап от метаболитното разграждане на тирозина протича с участието на специфичен ензим и образуването на междинно съединение.
И така, първо аминокиселината се разгражда до пара-хидроксифенилпируват, който се превръща в алкаптон – 2,5-дихидроксифенилоцетна или хомогентизинова киселина. След това алкаптонът би трябвало да се трансформира в малеоцетна киселина, но това не се случва. [ 8 ]
А патогенезата на алкаптонурията се състои в спиране на биохимичните реакции на катаболизма на тирозина на етапа на образуване на хомогентизинова киселина: просто няма ензим, необходим за нейното разграждане – хомогентизатоксидаза.
Хомогентизиновата киселина не се използва от организма и може да се натрупа чрез екскреция през бъбреците. Освен това, тя се окислява до бензохиноацетат (бензохинонооцетна киселина), който, чрез свързване с молекулите на тъканите и телесните течности, образува биополимерни съединения, оцветени като меланин.
Натрупването на тези междинни продукти в тъканта води до нарушаване на колагеновата структура на хрущялната тъкан, което намалява нейната еластичност – с появата на много клинични признаци на алкаптонурия и развитието на усложнения.
Симптоми алкаптонурия
Алкаптонурията при новородени и кърмачета се характеризира с потъмняване на урината. При контакт с въздух, урината върху пелени, памперси и бельо става тъмнокафява; това се дължи на натрупването и освобождаването на хомогентизинова киселина, която се окислява до бензохиноацетат. [ 9 ]
При липса на други симптоми, алкаптонурията при малки деца често не се разпознава своевременно, тъй като урината може да потъмнее след няколко часа уриниране. Според някои данни само една пета от децата под 12 месеца, родени с този ензимен дефицит, се идентифицират в клинични условия. Ето защо е много важно родителите да обръщат внимание на грижите за своите бебета.
Освен това, ранните признаци включват пигментация (синкаво-сив цвят) на склерата на очите и хрущялите на ушите и носа, което често се нарича охроноза.[ 10 ]
С течение на времето се появяват и други симптоми:
- силна пигментация на кожата по скулите, подмишниците и гениталиите;
- оцветяване на дрехите при контакт с потни участъци от тялото;
- пристъпи на обща слабост;
- дрезгав глас.
Трябва да се има предвид, че алкаптонурия и охроноза, както бе отбелязано по-горе, са синоними на едно и също нарушение на катаболизма на тирозина.
Кленов сироп, причинен от ушна болест, и алкаптонурия. Вродената кленов сироп, причинен от ушна болест, или левциноза, също е метаболитно разстройство, има същия модел на наследяване и дори мутации се появяват на една и съща хромозома, но засягат гена, кодиращ ензимния комплекс от разклонена верига α-кетокиселина дехидрогеназа. Поради това тялото не може да разгради определени компоненти на протеините, по-специално аминокиселините левцин, изолевцин и валин. При това заболяване урината (и ушната кал) имат сладък мирис; освен това клиничната картина на този вид органична ацидемия включва хипопигментация, колебания в кръвното налягане, гърчове, повръщане и диария, спадане на нивата на кръвната захар, кетоацидоза, халюцинации и др. Смъртността при децата е доста висока; при възрастни, без лечение, може да настъпи кома и смърт поради мозъчен оток.
Албинизмът и алкаптонурията са „обединени“ само от тирозин. Албинизмът, включително окулокутанният, се причинява от генетични мутации, които засягат производството на пигмента меланин. Вродени промени се наблюдават в гена TYR на хромозома 11 (11q14.3), който кодира тирозиназа, съдържащ мед меланозомен ензим, необходим за образуването на кожен пигмент въз основа на продукти от метаболизма на тирозин. Това заболяване е много по-често срещано от алкаптонурията.
Усложнения и последствия
Причинена от действието на междинните метаболити на тирозина – хомогентизинова и бензохинонооцетна киселини – последствията и усложненията на алкаптонурията се появяват поради отлагането на реактивни пигментирани полимери, разрушаването на колагеновите фибрили и влошаването на състоянието на хрущялите (с намаляване на тяхната устойчивост на механично натоварване).
С течение на годините, в зряла възраст, се развиват дегенеративни артрити и остеоартрити на големите стави (тазобедрени, сакроилиачни и колянни); междупрешленните пространства се стесняват (особено в лумбалната и гръдната част на гръбначния стълб) – с калцификация и образуване на остеофити; плътността на тъканта на субхондралните костни пластини намалява и подлежащите кости могат да претърпят патологично ремоделиране с образуване на израстъци и деформация. [ 11 ]
Може да се наблюдава увреждане на сърдечните клапи (аортна и митрална) и коронарните артерии – с признаци на коронарна болест на сърцето, както и образуване на камъни в бъбреците и простатната жлеза – поради същата калцификация. [ 12 ], [ 13 ]
Диагностика алкаптонурия
Обикновено диагнозата на вродени метаболитни нарушения се основава на изследване на биологичните течности на тялото.
Въз основа на какви тестове и реакции може да се диагностицира алкаптонурия? Необходими са изследвания на урината, за да се открие хомогентизинова киселина и да се определи нейното ниво (нормално – 20-30 mg на ден, повишено – 3-8 g). Проба от урина се изследва чрез газова хроматография или масспектрометрия, като се използва течна хроматография; възможен е скринингов тест за наличие на железен хлорид в урината. [ 14 ]
Съществува и метод за бърза диагностика – определяне на алкаптон в изсъхнали петна от урина върху хартия (чрез интензитет на цвета).
При изясняване на диагнозата, инструменталната диагностика (рентгенография) включва идентифициране на радиологични признаци на остеоартрит и други ставни патологии при пациенти.
Диагнозата се потвърждава чрез молекулярно-генетични методи за диагностициране на наследствени заболявания, като генетично изследване и ДНК секвениране. [ 15 ]
Диференциална диагноза
Диференциалната диагноза включва хемохроматоза и остра чернодробна недостатъчност на новороденото, меланинурия, остра интермитентна порфирия, хемофагоцитна лимфохистиоцитоза, първична митохондриална патология, ревматоиден артрит, анкилозиращ спондилит.
Към кого да се свържете?
Лечение алкаптонурия
Основното лечение за алкаптонурия е пероралното приложение на големи дози (поне 1000 mg на ден) аскорбинова киселина. При деца това увеличава екскрецията на хомогентизинова киселина в урината, а при възрастни намалява съдържанието в урината на нейното производно, бензохиноноцетна киселина, и забавя свързването ѝ със структурите на съединителната тъкан на ставите и колагена. [ 16 ]
Западноевропейските клиники тестват лекарството Нитизинон (Орфалин), лекарство от групата на метаболитите, което инхибира втория етап на катаболизма на тирозина: трансформацията на пара-хидроксифенилпируват в хомогентизинова киселина. Употребата на този фармакологичен агент обаче води до натрупване на тирозин и може да причини тежки странични ефекти, включително непрозрачност на роговицата и фотофобия, кървене от носа и стомаха, чернодробна недостатъчност, промени в кръвта и др. Въпреки това, в Съединените щати Нитизинон е одобрен от FDA за лечение на тирозинемия тип I. [17 ], [ 18 ]
Поради това, при проблеми със ставите, причинени от алкаптонурия, се провежда физиотерапевтично лечение – лечебна терапия за увеличаване на мускулната сила и подобряване на подвижността на ставите, балнеотерапия и пелоидна терапия за ограничаване на болката.
Въпреки че тирозинът не само се доставя с храната, но и се произвежда в организма, на пациентите с алкаптонурия се препоръчва да спазват нископротеинова диета и да ограничат консумацията на храни, богати на тирозин, предимно говеждо и свинско месо, млечни продукти (особено сирена), бобови растения, ядки и семена.
Предотвратяване
Предотвратяването на генните мутации е невъзможно, но за да се предотврати раждането на деца с висок риск от вродени нарушения, се провежда медицинско генетично консултиране, което е необходимо преди планирана бременност за двойките, чиято фамилна анамнеза включва наследствени заболявания. [ 19 ]
Прогноза
Фаталните последици от алкаптонурия са много редки и смъртта може да бъде причинена от сериозни усложнения, засягащи сърцето и бъбреците. Така че общата продължителност на живота на хората с алкаптонурия е добра.
Но качеството на живот се намалява поради интензивна болка в ставите или гръбначния стълб със значително ограничаване на подвижността, често прогресивна.