Медицински експерт на статията

Нови публикации
Синдром на Трише Колинс
Последно прегледани: 23.04.2024

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.

Когато нарушения феталното развитие на костите процеси имат сериозен краниофациална деформация, и една от разновидностите на такава патология е синдром Treacher Collins (СТК) или mandibulofastsialny, т.е. Лицево-челюстната дизостоза.
Кодекс на заболяването съгласно МКБ 10: клас XVII (вродени аномалии, деформации и хромозомни аномалии), Q75.4 - мандибуло-фациална дисстоза.
Причини на синдрома на Tricher Collins
Този синдром е кръстен изключителен британски офталмолог Едуард Treacher Колинс, който описва основните характеристики на заболяването преди повече от сто години. Въпреки това, лекарите в Европа често наричат този вид аномалии лицевите кости и челюстта заболяване или синдром Franceschetti - въз основа на задълбочени изследвания на швейцарската офталмолог Адолф Franceschetti, който въвежда термина "mandibulofastsialny дизостоза" в средата на миналия век. В медицинските среди се използва и името - синдром на Franceschetti-Collins.
Предизвиква синдром Treacher Collins - генни мутации TCOF1 (локус 5q31.3-33.3 хромозоми), който кодира nucleolar фосфопротеин, който е отговорен за формиране на краниофациална част на човешки ембрион. В резултат, преждевременно намали количеството на този протеин се разграждат биогенезата и функция на рРНК. Според генетиците изследователска програма човешкия геном, тези процеси водят до намаляване на разпространението на ембрионални невронни клетки CREST - ролката по нерв канавката, които по време на развитие на ембриона в невралната тръба се затваря.
Получаване на предната част на черепа тъкан се дължи на трансформация и клетъчна диференциация горната (глава) част на невралната тръба, които мигрират по продължение на невралната тръба към първата и втората с хриле арки ембриона. И дефицитът на тези клетки причинява краниофациални деформации. Критичният период на възникване на аномалии е от 18 до 28 дни след оплождането. След завършване на миграцията на невралната тръба клетки (в четвъртата седмица на бременността) са оформени почти всички свободно мезенхимни тъкан в лицето, което по-късно (5 до 8 седмици) се диференцират в скелетната и съединителната тъкан на всички части на лицето, шията, гърлото, ухото (включително вътрешни) и бъдещи зъби.
Патогенеза
Патогенезата на синдром Treacher Collins често се движи в семейства и аномалия се наследява по автозомно доминантен принцип, въпреки че има случаи на автозомни рецесивни предаване дефекти (мутации в други гени, по-специално, POLR1C и POLR1D). Най-непредсказуемите в лицево-челюстна дизостоза е, че мутацията е наследен от деца само в 40-48% от случаите. Това означава, че 52-60% от пациентите причинява синдром Treacher Collins не са свързани с наличието на аномалии в семейството и се смята, че има патология възниква от спорадични генетични мутации де ново. Най-вероятно новите мутации представляват ефектите от тератогенни ефекти върху плода по време на бременност.
Сред причините за този синдром тератогенни експертите наричат високи дози етанол (етилов алкохол), радиация, цигарен дим, tsitomegavirus и токсоплазмоза, както и хербициди глифозат (Raundal, Glifor, Tornado, и др.). Списъкът с йатогенни фактори включваше препарати за акне и себорея с 13-цис-ретиноева киселина (Isotretinoin, Accutane); антиконвулсивно лекарство фенитоин (дилантин, екпанутин); психотропни лекарства Diazepam, Valium, Relanium, Seduxen.
Симптоми на синдрома на Tricher Collins
В по-голямата си част клиничните признаци на мандибулофосалната дисостоза и степента на тяхната тежест зависят от характеристиките на проявата на генни мутации. И първите признаци на тази аномалия в повечето случаи се наблюдават в детето веднага след раждането му: лицето със синдрома на Трише Колинс има характерен външен вид. Освен това, морфологичните аномалии обикновено са двустранни и симетрични.
Най-очевидните симптоми на синдрома на Tricer Collins са:
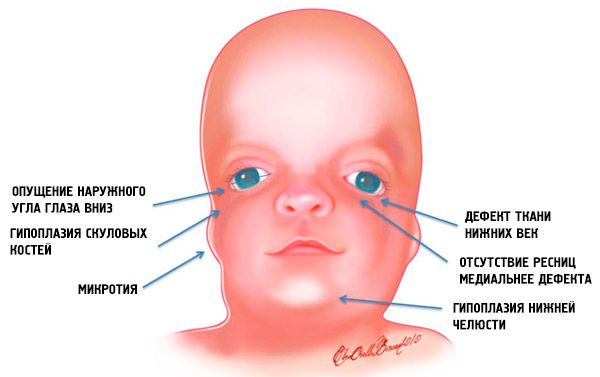
- недоразвитост (хипоплазия) лицевите кости на черепа: ябълчната, ябълчната процес на челната кост, страничните криловидни плочи, синусите, мандибуларния костни епифизи и издатини (кондилите);
- недостатъчно развитие на костите на долната челюст (микрогнатия) и по-тъп, отколкото обикновено ъгълът на мандибула;
- носът има нормален размер, но изглежда чудесно заради хипоплазма на горските арки и недостатъчното развитие или липсата на зигматични арки в района на храмовете;
- очните процепи са низходящи, т.е. Разрязването на очите е ненормално, като външните ъгли се спускат надолу;
- дефекти на долните клепачи (coloboma) и частично отсъствие на миглите върху тях;
- уши с неправилна форма с широк диапазон от отклонения, до тяхното положение в ъгъла на долната челюст, отсъствие на лобове, слепи фистули между козето ухо и ъгъла на устата и т.н .;
- стесняване или инфекция (атрезия) на външния слухов канал и аномалии на осикулите на средното ухо;
- отсъствие или хипоплазия на паротидните слюнчени жлези;
- фарингеална хипоплазия (стесняване на фаринкса и дихателните пътища);
- нелечение на твърдото небце (вълна на устата), както и отсъствието, съкращаването или неподвижността на мекото небце.
Такива анатомични аномалии във всички случаи имат усложнения. Това са функционални слухови увреждания под формата на проводими (проводими) слухови загуби или общо глухота; зрително увреждане поради неправилно формиране на очните топки; Дефектите на небцето причиняват затруднения при хранене и преглъщане. Има нарушения на челюстта на запушването на зъбите (неправилна захапка), което на свой ред причинява проблеми с дъвченето и артикулирането. Патологиите на мекото небце обясняват носния глас.
Усложнения и последствия
Последиците от черепно-лицеви аномалии в синдром Treacher Колинс проявява в това, че при раждането нормалното си интелектуални способности, но поради увреждане на слуха и други нарушения, отбелязани вторичен умствена изостаналост.
Освен това децата с такива дефекти осъзнават остро своята малоценност и страдат, което отрицателно засяга тяхната нервна система и психика.
Диагностика на синдрома на Tricher Collins
Постнаталната диагноза на синдрома на Tricher Collins се основава основно на клинични признаци. Долностолната дисостоза се определя лесно с пълна изразителност на синдрома, но когато има минимални симптоми на патологията, могат да възникнат проблеми при формулирането на правилната диагноза.
В този случай е необходимо специално внимание, за да се оценят всички функции, свързани с аномалии, особено тези, които засягат дишането (поради заплахата от сънна апнея). Извършва се и оценка и мониторинг на ефективността на хранене и насищане на хемоглобина с кислород.
В бъдеще - на 5-6-ия ден след раждането - е необходимо да се установи степента на увреждане на изслушването с помощта на аудиологични тестове, които трябва да се извършват в майчинството.
Извършва се преглед, при който инструменталната диагноза се извършва чрез флуороскопия на черепно-лицевата дисморфология; пантомография (панорамно рентгеново изследване на костни структури на лицевия череп); пълна черепна компютърна томография в различни проекции; CT или MRI на мозъка, за да се определи състоянието на вътрешния слухов медус.
Най-ранната - Пренатална - диагностика на черепно-лицеви аномалии в присъствието на синдром Treacher Колинс, фамилна анамнеза е възможно чрез хорионбиопсията 10-11 седмица от бременността (процедурата заплашва спонтанен аборт и инфекция в матката).
Също така се правят кръвни изследвания на членове на семейството; на 16-17 седмици от бременността се взема анализ на амниотична течност (трансабдоминална амниоцентеза); на 18-20 седмици от бременността се извършва фетоскопия и се взема кръв от плодовите съдове на плацентата.
Но най-често в пренаталната диагноза на този синдром плода използва ултразвук (на 20-24 седмици от бременността).
Какви тестове са необходими?
Диференциална диагноза
Тези същите методи експерти използват, когато е необходимо диференциална диагноза, да се признае леко изразен синдром Treacher Collins и се разграничи от други вродени аномалии на черепно-лицеви кости, по-специално: синдроми Apert, Crouzon, Nagera Peters-Hevelsa, Hellerman-Staefa, както и с лицев microsomia (синдром Goldenhar), хипертелоризъм, началото неперфорирани черепните шевове (craniostenosis) или нарушение на сливане на лицевите кости (краниосиностоза).
Лечение на синдрома на Tricher Collins
Както във всички случаи на генетично обусловени вродени дефекти, лечението на синдрома на Tricer Collins в тежки форми е изключително палиативно, тъй като просто няма терапевтични методи за такива патологии. Спектърът и степента на деформация при този синдром са обширни и следователно естеството и интензивността на медицинската интервенция има и разнообразие от възможности.
Слуховите апарати се използват за коригиране и подобряване на слуха, за подобряване на лекциите с реч терапевт.
Хирургична интервенция е необходимо в ранна възраст, в тежки случаи на дихателните пътища стесняване (трахеостомия се извършва) и ларинкса (извършва хранене gastrostommiya) може да изисква хирургична корекция на небцето.
Операциите за удължаване на долната челюст се извършват на възраст 2-3 години или по-късно. Реконструкцията на меките тъкани включва корекция на долните колонии на клепачите и пластиката на ушите.
Прогноза
Какво може да бъде прогнозата за тази патология? Това зависи от степента на деформация и интензивността на симптомите. Синдромът Трикър Колинс е диагноза за цял живот.
 [25]
[25]