Медицински експерт на статията

Нови публикации
Синдром на Angelmann при деца и възрастни
Последно прегледани: 23.04.2024

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.

Има редица заболявания, при които изрази като "се грижи за себе си и не се разболявай" звучат поне нелепи. Тази патология, при която някои психически и физически аномалии са вградени в тялото на детето още преди раждането, но родителите нямат вина. Такива заболявания се причиняват от мутации или нарушения в хромозомните комплекти и се наричат хромозомни или генетични. Синдромът на Ангелман, синдромът на Даун, Патау, Едуардс, Търнър, Прадер-Уили са само част от генетичните заболявания от доста приличен списък.
Синдром на щастлив човек
Този път ние говорим за болестта, наречена на името на британския педиатър Хари Angelman от тази първа повдигна въпроса за проблем 1965 година, пред които са изправени в навечерието на своята практика с три необичайни деца, обединени от общи характерни симптоми. Лекарят нарече тези деца кукленски деца и написа за тях статия, която първоначално беше наречена "Кукленски деца". Самата статия и името й са написани под впечатлението на картина, която се вижда в един от музеите във Верона. Картината изобразяваше смешно момче и се казваше "Кук-марионетка". Асоциацията е показано на снимката на детето с трите деца с Angelman който веднъж се сблъскват в практиката си и избута педиатър деца комбинират в една група, защото на съществуващите им състояние.
Фактът, че децата в статията не са забелязани от други лекари, не е изненадващо. В края на краищата на пръв поглед изглеждаше, че имат съвсем различни заболявания, така че общата клинична картина на заболяването се различава в 3 различни случая. "Нова" хромозомна патология може да е от интерес за други учени, но по това време генетиката все още не е достатъчно развита, за да потвърди хипотезата на английски лекар. Следователно статията след определен интерес към нея отдавна е изоставена в далечния полк.
Следващото споменаване на синдрома на Angelman, и така се нарича статия на педиатър от Англия, Г. Англеман, датира от началото на 80-те години на ХХ век. И едва през 1987 г. Е възможно да се намери причината, поради която малка част от децата се раждат с такива отклонения, че отстрани те изглеждат постоянно усмихнати и щастливи. Всъщност това не е така, а усмивката е само гримаса, зад която се крие нещастната човешка душа и болката на родителите.
Епидемиология
Хромозомната мутация при дете, според статистиката, може да се развие както на фона на такива мутации в родителите, така и при отсъствието на такива. Няма ясен наследствен характер при синдрома на Angelmann (CA), но вероятността за развитие на патология при родители с хромозомни мутации е доста висока.
Интересно е също така, че ако едно семейство вече има дете със СА, има шанс от един процента да има второ дете от същия тип, дори ако родителите са здрави.
Все още няма точна статистика за броя на пациентите със синдром на Anghelman. Може би вината е разнообразие от симптоми, които могат да се появят в определен състав или за дълго време въобще не възникват. Предполага се, че разпространението на заболяването е: 1 дете на 20 000 новородени. Но тази цифра е много приблизителна.
Причини синдром на ангелман
Синдромът на Ангелман е медицинското име за хромозомна патология, но в никакъв случай не е единственият. При хората това заболяване се нарича синдром на марионетки и синдром на щастлива марионетка, синдром на Петрушка и синдром на смешна кукла. Да, какви имена хората не могат да измислят (понякога дори обиждат за самите пациенти и техните родители), но болестта е болест, колкото и странно да изглежда външно и каквито и да са причините за това.
И причините за развитието на синдрома на Angelman, както и много други генетични патологии, във всички случаи са нарушения в структурата на един от хромозомите или хромозомния набор като цяло. Но само в нашия случай целият проблем е в 15-те хромозоми, предавани от майката. Т.е. Бащината хромозома в този случай няма отклонения, но жената претърпява определени мутации.
Според вида хромозомни аномалии синдромът на Angelmann се отнася до хромозомни мутации. Такива мутации са:
- Заличаване (липса на хромозома регион съдържащ специфичен набор от гени, и ако нито един от гените става въпрос микроделеции), което е резултат от две прекъснатости и един събиране когато губи част от оригиналната хромозомата.
- Дублирането (наличието на допълнителен сайт в хромозомата, което е копие на вече наличното), което в повечето случаи води до смърт на човек, по-рядко - до безплодие.
- Инверсия (инверсия на хромозома секции на 180 градуса, т.е., в обратна посока, и след това в него гени са подредени в обратен ред), когато прекъснатата хромозомата краища свързани в ред, различен от оригинала.
- Вмъкване (ако част от генетичния материал в хромозомата не е на мястото му),
- транслокация (ако част от хромозомата се свързва с друга хромозома, такава мутация може да бъде взаимна без загуба на места).
Получавайки мутирала хромозома от нищо неподозираща майка, бебето е обречено да се роди с отклонения предварително. Най-честата причина за развитието на синдрома на Ангелман все още е изтриването на майчината хромозома 15, когато в нея няма малка площ. По-рядко срещащи се мутации в синдрома на "смелата кукла" са:
- преместване,
- диозомия с един баща (ако детето получи чифт хромозоми от бащата, липсва майчината хромозома)
- мутация на гени в ДНК, които са и основен материал (генетичен материал) и инструкция за правилното му използване (по-специално мутация на ube3a гена в майчината хромозома).
Наличието на една от тези мутации в родителите е рисков фактор за синдрома на Anghelman при деца. Но не само хромозомните мутации, но и геномните мутации (които са свързани с количествена промяна в хромозомните комплекти и се срещат по-често от хромозомите) могат да предизвикат развитието на болестта при детето. За общи геномни мутации може да се отдаде тризомия на хромозомите (ако човек има хромозомен набор има повече от 46 хромозоми).
За патологията на бебето не е задължително родителите да имат хромозомни аномалии. И все пак има определен процент пациенти, чието заболяване е наследствено.

Патогенеза
Нека да копаем малко в биологията, по-точно в генетиката. Генетичната информация за всяко отделно човешко тяло се съдържа в 23 двойки хромозоми. Една хромозома от двойката се предава на детето от бащата, а другата от майката. Всички двойки хромозоми се различават по форма и размер и носят в себе си определена информация. Така че 23 двойки хромозоми (X и Y хромозоми) са отговорни за формирането на сексуалните характеристики на детето (XX - момиче, XY-boy, докато едно дете може да получи Y-хромозома само от баща си).
В идеалния случай детето получава от родителите си 46 хромозоми, които формират неговите генетични атрибути, които го определят като индивид. По-голям брой хромозоми се нарича тризомия и се счита за отклонение от нормата. Например, наличието на 47 хромозома в хромозомния набор (кариотип, който определя видовете и индивидуалните характеристики) предизвиква появата на синдрома на Даун.
Ако хромозомите са оцветени със специално боядисване, тогава в микроскопа могат да се видят ленти с различни нюанси по всеки от тях. Във всяка група има огромен брой гени. Всички тези групи са преброени от учените и имат фиксирано местоположение. Липсата на една от лентите се счита за отклонение от нормата. При синдрома на Angelmann много често наблюдавате липсата на сегменти от майчината хромозома в интервала q11-q13, намиращ се в дългата ръка, броят на ДНК базите, в които са само около 4 милиона.
Основният компонент на хромозомата е невероятно дълга ДНК молекула, съдържаща хиляди гени и десетки и стотици милиони азотни бази. По този начин, 15-те хромозоми, отговорни за развитието на синдрома на Angelman и няколко други, съдържат 1200 гени и около 100 милиона бази. Всяко нарушение в структурата на ДНК молекулата задължително ще повлияе на появата и развитието на нероденото дете.
Генетичната информация, съдържаща се в гените, се превръща в протеин или РНК. Този процес се нарича генна експресия. По този начин генетичната информация, получена от родителите, получава както форма, така и съдържание, въплътено в уникалния им наследник на женския или мъжкия пол.
Има редица патологии с некласически тип наследство, включително Angelman синдром, при който гените, получени от родители като част от сдвоени хромозоми са уникален печатен родители и се проявяват по различен начин.
Така че, Angelman синдром е отличен пример на геномна отпечатване, при което експресията на гени в тялото на детето е пряко в зависимост от които майки получен алели (са разположени на еднакви части на сдвоени хромозоми различни форми на същия ген, получен от бащата и майката) , Т.е. Да доведе до появата на синдром, нарушения в хромозомата на майката, а мутации и разстройства бащините хромозомната структура предизвикват много различни заболявания.
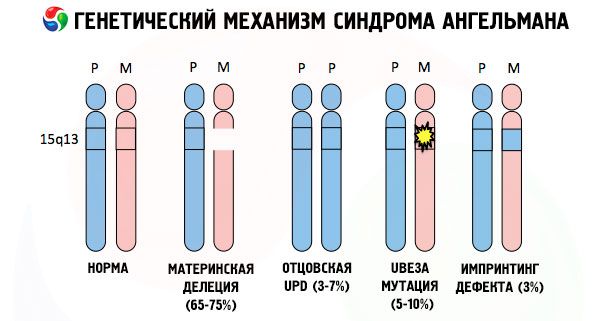
При това заболяване има липса на специфични гени в хромозомата на майката или загуба / намаление на активността на отделните гени (в повечето случаи ube3a ген, включен в метаболизма на убиквитин - протеиново разграждане други регулаторни протеини). Вследствие на това детето се диагностицира с аномалии в умственото развитие и физически деформации.
Симптоми синдром на ангелман
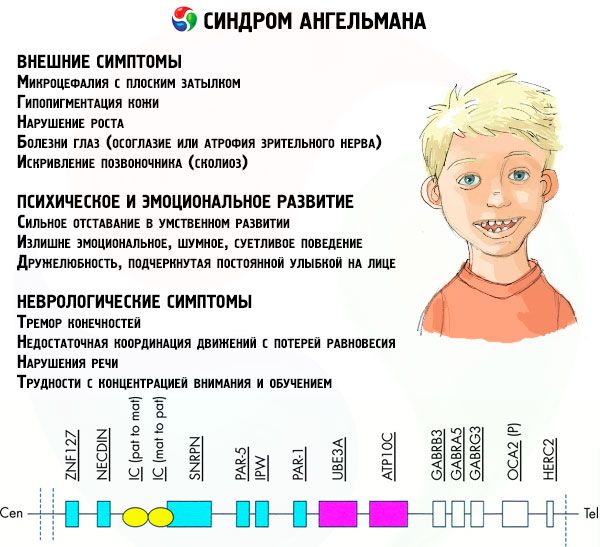
Симптомите на синдрома на Angelman засягат различни аспекти на живота и развитието на детето: физически, неврологични и психически. Въз основа на това можем да различим три групи симптоми, които показват развитието на тази патология.
- Външни или физически симптоми:
- несъразмерно малка глава в сравнение с багажника и крайниците, които са с нормален размер,
- твърде широка уста,
- на лицето почти винаги има усмивка (с отворена уста),
- редки зъби,
- тясна горна устна,
- често залепване на широк език,
- изпъкнала долна челюст,
- остра брадичка,
- много лека кожа, често и косми (албинизъм, свързан с факта, че тялото не произвежда пигмента меланин),
- тъмни петна по светлата кожа (хипопигментация поради недостатъчно производство на меланин)
- физически или външни симптоми: очни заболявания като страбизъм или атрофия на оптичния нерв,
- кривина на гръбначния стълб (сколиоза),
- твърди крака (когато ходене човек не огъва коленете си поради малката подвижност на ставите, оттук и сравнението с марионетната походка).
- Симптоми, свързани с психическо и емоционално развитие:
- силно изоставане в психическото развитие,
- ненужно емоционално, шумно, раздразнено поведение,
- често пляскане,
- изразената привързаност, подчертана от постоянната усмивка на лицето,
- често, безполезен смях.
- Неврологични симптоми:
- тремор на крайниците,
- недостатъчна координация на движенията със загуба на равновесие,
- намален мускулен тонус,
- различни разстройства на съня,
- чести истерични припадъци в детска възраст,
- увреждания на говора (детето започва да говори късно, има лоши умения за комуникация и неразбираема реч),
- хиперактивност на фона на повишена възбудимост,
- трудности при концентрация и обучение.
Но това е обобщена картина на болестта. Всъщност, клиничната картина на синдрома на Anghelman до голяма степен зависи от стадия на развитие на заболяването и вида на хромозомната мутация, причиняваща патологията. И това означава, че при различни пациенти симптоматиката на заболяването може да се различава значително, което за дълго време не позволява изолиране на патологията, наред с други, с подобна клинична картина.
От общия брой симптоми могат да бъдат идентифицирани такива, които са характерни за всички пациенти без изключение:
- тежки отклонения в умственото развитие,
- неадекватно поведение (безсмислен смях, повишена възбуда, слаба концентрация на внимание, състояние на еуфория);
- недостатъчно развитие на двигателните умения,
- лоша координация на движенията, атаксия на ходене (неравномерно темпо, люлеене от страна на страна и т.н.), тремор на крайниците.
- нарушаване на речта с преобладаване на невербалните средства за комуникация.
Сред симптомите, които се наблюдават при по-голямата част от пациентите, можем да различим тези:
- несъразмерна глава и багаж, причинени от забавяне на физическото развитие,
- при много пациенти формата на черепа е такава, че размерът на мозъка остава по-малък, отколкото при здрави хора (микроцефалия)
- епилептични припадъци на възраст до 3 години с прогресивно намаляване на силата и честотата при пациенти в старческа възраст,
- изкривяване на индексите на ЕЕГ (колебания и висока амплитуда на нискочестотните вълни).
Тези симптоми се срещат доста често, въпреки това при 20% от пациентите със синдром на Angelmann те отсъстват.
Още по-рядко може да се диагностицират такива прояви на болестта като:
- изразен или лек страбизъм,
- слаб контрол върху движението на езика, в резултат на което пациентите често избиват езика си без причина,
- трудности при преглъщане и смучене, особено при по-малки деца,
- нарушение на пигментацията на кожата и очите,
- повдигнати или огънати по време на ходене на ръце,
- гиперрефлексия,
- нарушения на съня, особено в детството,
- често слюнчване,
- неудържима жажда,
- прекомерно активни дъвчащи движения,
- свръхчувствителност към топлина,
- плоска глава,
- напреднала долна челюст,
- плавни палми.
Много голям процент от пациентите имат проблеми с уринирането, които лошо контролират, нарушаване на фините двигателни умения, което създава трудности при самообслужване и обучение, с наднормено тегло. Практически при всички пациенти пубертетът започва по-късно, отколкото при здрави връстници.
Децата със синдром на ангелман са добри в разбирането и разбирането на устната реч, но не искат да участват в разговора, като ограничават речта си до десетки думи, необходими в ежедневието. Но в зряла възраст такива пациенти изглеждат по-млади от техните връстници без генетични патологии.
Много от симптомите на синдрома на Ангелман са непостоянни, така че клиничната картина на болестта се променя с възрастта. Припадъците и епилептичните припадъци стават все по-редки или изобщо изчезват, пациентът става по-малко възбуден, сън се приготвя.
Усложнения и последствия
Синдромът на Angelman е сериозна, практически неизлечима до момента хромозомна патология, която лишава пациентите от възможността да живеят нормално. Какъв ще бъде животът на бебето с SA, до голяма степен зависи от вида на хромозомната аномалия.
Дублирането на хромозомната област в повечето случаи е несъвместимо с живота. И дори тези пациенти да не умират в ранна детска възраст и да не стигат до пубертета, те нямат възможност да имат деца.
Заличаването или отсъствието на част от гените, които се появяват най-често при синдрома на Anghelman, е пречка за детето да се учи да ходи и да говори. При такива деца, умственото забавяне е представено в по-тежка форма, често се появяват атаки на епилепсия и интензивността им е много по-силна, отколкото при пациенти с други хромозомни аномалии.
Ако има мутация на един-единствен ген, като се отчита надлежно и подход, детето може да се научи на основите на комуникация самообслужване и комуникация в екипа, въпреки че все още ще изостава в развитието на своите връстници.
За децата със синдром на Ангелман, доброжелателни по природа, основната е любовта и вниманието на родителите. Само в този случай обучението на детето ще донесе плодове, дори малки. Разбира се, пациентите в редовното училище няма да могат да учат със спортната академия. Те се нуждаят от специални класове, в които децата първо ще се научат да съсредоточават вниманието си, а след това постепенно ще дадат основите на училищното знание.
Диагностика синдром на ангелман
Синдромът на Angelman е вродена патология на развитието. Но поради някои обстоятелства, диагностицирането в ранна детска възраст и детството най-често не е възможно. Причината за това е неспецифичността и леките симптоми при кърмачета и малки деца до 3 години. А разпространението на болестта в нашата страна не е толкова голямо, че лекарите са се научили да я разпознават сред нейните.
Angelman синдром при новородени може да се прояви под формата на намаляване на мускулния тонус, което се проявява под формата на проблеми с храненето (слабост смучещи и гълтане рефлекс), и по-късно затруднения в обучението, да ходят (такива деца много по-късно започват ходене). Тези симптоми са първите признаци на отклонение в развитието на бебето, което може да бъде свързано с хромозомна аномалия. Потвърдете това предположение може само генетичен анализ.
Особено внимание се обръща на деца, чиито родители имат различни геномни или хромозомни аномалии. След popervosti заболяване не може да се прояви по някакъв начин, и ако се открият аномалии във времето, започна да се енергично се ангажират с детето, то е възможно да се постигне много по-голям успех в обучението, блокиране на прогресията на заболяването.
Ако родителите имат различни хромозомни аномалии, генетичният анализ се извършва още преди раждането на бебето, тъй като СА е една от патологиите, които могат да бъдат открити в ембрионално състояние.
Събирането на материали за генетични изследвания може да се извърши по два начина:
- инвазивен (с определен процент риск, тъй като се изисква да влезе в матката, за да вземе тест за амниотична течност),
- неинвазивен (ДНК анализ на бебето от кръвта на майката).
След това се извършват следните изследвания:
- флуоресцентна in situ хибридизация (FISH метод) - свързване на ДНК сонда, белязана със специално оцветител към изследваната ДНК, последвано от микроскопско изследване.
- анализ на мутации в ube3a гена и отпечатване на гени,
- анализ на ДНК метилирането с помощта на специални методи, използвани в генетиката.
Генетичните анализи дават доста точна информация в случай на хромозомни аномалии, така че бъдещите родители предварително да знаят за какво трябва да бъдат подготвени. Въпреки това има изключения. При определена група пациенти, при наличието на всички симптоми, показващи симптомите, резултатите от анализите остават нормални. Т.е. За да се разкрие патологията, е възможно единствено внимателно да се наблюдава детето от най-ранното детство: как се храни, когато той започва да ходи и да говори, дали краката се навеждат при ходене и т.н.
В допълнение към FISH-метод, сред методи за диагностично средство Angelman синдром могат да бъдат разграничени томография (СТ или MRI), което помага да се определи състоянието и размера на мозъка, и електроенцефалограмата (ЕЕГ), което показва, как отделните части на мозъка на труд.
Окончателната диагноза на лекарите обикновено се определя на възраст 3-7 години, когато пациентът вече има повечето симптоми и динамиката на развитието на болестта е видима.
Какви тестове са необходими?
Диференциална диагноза
Синдромът на Angelman е генетична патология, която всъщност няма специфични прояви. Повечето от симптомите могат да показват както CA, така и други генетични патологии.
Диференциалната диагноза при синдрома на Anghelman се извършва със следните патологии:
- Синдром на Пит-Хопкинс (пациентите се характеризират с умствена изостаналост, весел характер, усмихнат, те имат доста голяма и широка уста, се забелязва микроцефалия). Разлика - атаки на хипервентилация и забавяне на дишането в състояние на будност.
- Синдромът Kristiansona (пациентите с умствена изостаналост с весело разположение, неспособни да говорят, се характеризират с микроцефалия, атаксия, конвулсии, принудителни движения на мускулите).
- Синдром на Mowata-Wilson (симптоми: умствено изоставане, епилептични припадъци, остра брадичка, отворена уста, израз на щастие на лицето, микроцефалия). Разликата е голямо разстояние между очите, очите са скосени навътре, върхът на носа е заоблен, ушите са обърнати назад.
- синдром на Кабуки (характеризира се с лека до умерена степен на умствена изостаналост, проблеми с говора и двигателните умения, мускулна слабост, припадъци, микроцефалия, големи празнини между zudami, некоординираност). Разлика - вежди във формата на дъга, обърната странична част на долния клепач, широко разположени очи, дълги очни процепи с дълги гъсти мигли.
- Rett синдром (диференциация с СА при жените). Симптоми: забавено развитие на говора, конвулсивни припадъци, микроцефалия. Разликата - няма щастлив израз на лицето, има атаки на апнея и апраксия, които в крайна сметка прогресират.
- Синдром на автозомно-рецесивно умствена tardatsii 38 (симптоми: умствена изостаналост с забележимо забавяне в развитието на двигателните умения и речта, мускулна слабост, проблеми с храненето в ранна детска възраст, импулсивност). Разликата е синият цвят на ириса.
- Синдром на дублиране на ген MESR 2 (диференциация с SA при мъже). Симптоми: тежко умствено изоставане, мускулна слабост от детството, проблеми с говора или липса на такава, епилепсия. Разлики - прогресивна миопатия, постоянно повтарящи се инфекции.
- синдром Klifstra (симптоми: проблеми с говора и мислене, мускулна слабост, нарушения на съня, липса на внимание, отворена уста, възбуда, гърчове, атаксия, нарушено равновесие). Различия - плоско лице, къси нокти, широки очи, голяма обърната долна устна, атаки на агресия.
- Синдромът Смит-Магенис (характеризиращ се с гърчове, проблеми със съня, нарушения на интелектуалното и двигателното развитие). Разлики - широко и плоско лице, изпъкнало чело.
- Синдром на Kulena-de Vries (леко и умерено умствено изоставане, мускулна слабост, конвулсивни атаки, удобство). Разлики - дълго лице с високо чело, изпъкнали уши, наклонени очи, по-голяма мобилност на ставите, вродени сърдечни патологии.
- Синдром Филан - Макдермид (симптоми: умствена изостаналост, нарушение на говора или липса на такъв). Разлики - големи ръце с развити мускули, мускулна слабост от раждането, слабо изпотяване.
Angelman синдром подобни симптоми могат да "похвали" и как такива adenilsuktsinazy дефицит патология, автозомно-рецесивно синдром на умствена изостаналост 1, синдрома на дублиране хромозома 2q23.1, хаплоинсуфициенция гени FOXG1, STXBP1 или MEF2C и др.
Задачата на лекаря е да направи точна диагноза, да различава синдрома на Angelmann от патологии с подобни симптоми и да предпише ефективно лечение, което е от значение за диагностицираната степен на развитие на болестта.
Лечение синдром на ангелман
Синдромът на Ангелман се отнася до категорията на тези патологии, търсенето на ефективно лечение на коя лекарство е ангажирано до този ден. Етиологичното лечение на болестта е в етапа на разработване на различни методи и средства, много от които все още не са изследвани при хора. Досега лекарите трябва да бъдат ограничени до симптоматична терапия, за да помогне по някакъв начин облекчаване на положението на децата и възрастните със синдром на куклен, страда от епилептични припадъци, саливация, хипотония, както и нарушения на съня.
Така че, намалете честотата и силата на епилептичните припадъци може да бъде с правилно подбран антиконвулсант. Но проблемът е, че пристъпи при пациенти с СА са различни от обикновените епилептични припадъци, така че те се характеризират с няколко типа на припадъци, и по този начин да облекчи състоянието ще бъде въвеждането на няколко лекарства.
Най-популярни антиконвулсанта използва за лечение на Angelman синдром са: валпроева киселина, топирамат, ламотрижин, леветирацетам, клоназепам и препарати на базата на него. По-рядко се използват лекарства, базирани на karmazepina, фенитоин, фенобарбитал, етосуксимид, тъй като някои от тях може да предизвика парадоксален ефект, е да се засили и увеличаване на честотата на епилептични припадъци. Това се случва, ако лекарството се използва като част от монотерапията.
За лечение на слюноотделяне обикновено се използват два метода: медицински (препарати, потискащи образуването на слюнка) и оперативни, състоящи се в повторно имплантиране на слюнчените канали. Но в случая с CA, тези методи се считат за неефективни и въпросът остава отворен. Родителите и тези, които се грижат за такива пациенти, трябва да обърнем специално внимание на този момент, тъй като самите пациенти обикновено не контролират слюноотделяне, а някои просто не могат да се грижат за себе си.
Друг проблем е кратката продължителност на съня. Често децата със синдром на Ангелман спят не повече от 5 часа, което оказва негативно влияние върху работата на целия организъм. Стремежните, активни деца, любовните игри и комуникацията (дори ако се опитват да се ограничат до невербални начини), са забележимо уморени за деня. За да си почине добре, тялото се нуждае от здрав пълноценен сън, но това е само проблемът с него.
Тя ще изглежда, за да се подобри съня в възбудими пациенти трябва да бъде достатъчно лекарства с седативен ефект (фенотиазини и атипични антипсихотици), успокоява нервната система. Но в случая с CA, употребата на такива лекарства е изпълнена с появата на негативни ефекти. Следователно лекари предпочитат още леки хипнотични лекарства, такива като "Мелатонинът" (естествен хормонален препарат на основата на сън хормон), което даде пациенти час преди лягане в една таблетка, и "Дифенхидрамин". Честотата на прилагане и дозировката на пациента се определят от лекаря, в зависимост от състоянието и възрастта на пациента.
Понякога пациентите с синдром на ангелман имат проблеми с храносмилането и изпражненията. Регулирането на стол е възможно чрез лаксативни препарати (това е по-добре от фитогенеза).
И вие може да подходи към проблема по различен начин, както направи на американските лекари, въз основа на някои от методите за лечение на аутизъм, защото много от симптомите, характерни за SA, също са характерни за аутизма (импулсивност, неволни движения повтарящи се действия, дефицит на вниманието, проблеми в комуникацията и т.н. ) .. Установено е, че приложението на секретиновите хормона, нормализира храносмилането и стол, има положителен ефект върху вниманието и окситоцин пациентите помага за подобряване на когнитивните способности и паметта на децата, за да коригирате поведението.
Вярно е, че някои хормони са незаменими тук, особено когато става дума за деца. Синдромът на Ангелман показва поведенческа терапия, работа с психолог и логопед (преподаване на несловесни начини на комуникация и жестомимичен език). Обучението на такива деца трябва да се основава на индивидуална програма с участието на специално обучени учители, психолог и родители. За съжаление, това не е възможно навсякъде и семействата остават сами с проблема си.
Тъй като много малки пациенти с СА страдат от нисък мускулен тонус и проблеми със ставите, много внимание се отделя на физиотерапевтичното лечение. Най-често лекарите прибягват до използването на парафинови приложения, електрофореза, магнитотерапия.
Активен тонизиращ масаж и специални упражнения за физиотерапия ще помогнат на болното дете след известно време да стои уверено на краката си и да ходи. Особено полезно в това отношение аква-анимацията, която се препоръчва в CA в хладка вода. Повишава тонуса на мускулите и учи бебето да притежава своето тяло, координира движенията.
Антиконвулсивно лечение
Най-опасният симптом при синдрома на Anghelman е пристъпите, подобни на епилептичните припадъци. Този симптом се наблюдава при 80% от пациентите, което означава, че на всички от тях трябва да се предпише ефективно антиконвулсивно лечение.
Лечението на епилептичните припадъци се извършва с помощта на витамини и антиконвулсанти. Когато Angelman синдром, придружен от конвулсивен синдром, ще бъде полезно витамини B група, както и витамин С, D и Е. Но витамин терапия да определи собствените си в този случай е много опасно, тъй като неконтролирана прием на витамини може да намали ефективността на анти-епилептични лекарства и провокира нов, по-тежка и продължителна атаки.
Изборът на антиконвулсанти и назначаването на тяхната ефективна доза трябва да се извършват и от специализиран лекар. Той също така решава дали ще има достатъчно едно лекарство, или пациентът ще трябва да приема 2 или повече лекарства за дълго време .
Повечето пациенти лекари предписват лекарства валпроева киселина ( "валпроева киселина", "Depakinum", "Konvuleks", "valparin" и др.), Които пречат на конвулсии, подобряване на настроението и умствено състояние на пациентите.
Валпроевата киселина се предлага под формата на таблетки, сироп и инжекционни разтвори. Най-популярното лекарство е лекарството с продължително действие "Depakin" в таблетки и като разтвор за интравенозно приложение. Дозата на лекарството се определя от лекаря поотделно в зависимост от теглото, възрастта и състоянието на пациента.
Вземете лекарството по време на хранене 2 до 3 пъти на ден. Средната дневна доза е 20-30 mg на 1 килограм от теглото на пациента, максимумът е 50 mg / kg на ден.
Противопоказания за употреба. Той не се използва за нарушения на черния дроб и панкреаса, хеморагичен диатеза, хепатит, порфирия и свръхчувствителност към лекарството.
Сред нежеланите реакции могат да се разграничат треперене на ръцете, храносмилане и изпражнения, промени в телесното тегло.
"Топирамат" също е лекарство по избор в Калифорния. Той се произвежда под формата на таблетки и се използва като част от монотерапията и в комбинация с други лекарства.
Начин на приложение и дозировка. Вземете хапчета вътре, без оглед на приема на храна. Началният дневен прием за възрастни е 25-50 mg, за деца 0,5-1 mg / kg. Всяка седмица дозата се увеличава според предписанието на лекаря.
Лекарството не трябва да се приема по време на бременност и кърмене, както и с повишена чувствителност към неговите съставки. Лекарството има много различни странични ефекти.
Лекарства, които лекарят може да предпише на Angelman синдром "Klomazepam", "Rivotril" "Ламотрижин", "Seyzar", "Lamictal", "леветирацетам", "Keppra", "Epiterra" и др.
Алтернативно лечение и хомеопатия
Алтернативната медицина, като хомеопатичните лекарства, определено различава сравнителната безопасност, но тук ефективността на подобно лечение по отношение на синдрома на Angelholm може да се счита за противоречива.
Въпреки че някои алтернативни лечения все още могат да помогнат. Става въпрос за спиране на епилептичните припадъци. В това отношение билкова терапия може да бъде доста ефективна.
Добър ефект се постига чрез медицинска такса, основана на божур, сладник и кедрово дърво (компонентите се вземат в еднаква степен). Тревите трябва да се смилат на брашно. След 2 седмици от началото на приема, можете да забележите значително намаляване на честотата на конвулсивните атаки.
Полезно за спазми и отвара от лавандула (1 ч.л. За чаша вряща вода). Формулировката се вари 5 минути и се оставя за половин час. Вземете лекарството през нощта в продължение на 14 дни.
Ефективен при епилептични припадъци се счита за водна (или алкохолна) инфузия майчинство.
От хомеопатията лекарства за предотвратяване на гърчове с Angelman синдром може да се използва наркотици, чрез лайка и motherwort, Acidum hydrocyanicum, Argentum Nitricum, Калий bromatum, Arsenicum албум. Но е необходимо да се има предвид, че ефективните и безопасни дози от препарати във всеки конкретен случай могат да назначат само лекар хомеопатик.
Предотвратяване
Както вероятно е разбрал читателят, за да предотврати мутацията на гени и други хромозомни аномалии, медицината все още е извън силата, както и да се поправи ситуацията. Това може да се случи на всички, защото децата със синдром на Ангелман се раждат и при здрави родители, а генетиката, която в момента е един от най-слабоучилищните клонове на медицината, все още не може да обясни това.
Единственото, което може да се направи, е да поеме отговорността за планиране на бременността, да бъде регистрирана и проверена навреме. Но отново такава мярка по-скоро не би била профилактична, а когнитивна, както всяко изследване. Но младите родители предварително ще знаят за какво да се подготвят, а в случай на положителен отговор те ще решат дали ще могат да поемат такава отговорност като отглеждането на болно дете.
Прогноза
Прогнозата за синдрома на Anghelman зависи от характера на хромозомната аномалия и навременността на откриването му. Най-трудната част е за тези деца, чиито 15 хромозоми съдържат "липсващи" гени (заличава се). Вероятността за ходене и говорене при такива пациенти е изключително малка. Останалите случаи с внимателен подход и любов към вашето дете подлежат на корекция.
Такива пациенти, уви, не могат да станат пълноправни членове на обществото, въпреки факта, че те са далеч от глупави, разбират речта и значението му. Ето само проблеми с комуникацията, която те имат за живота. Пациентите могат да бъдат преподавани на жестомимичен език от детството, но човек не може да бъде принуден да комуникира с думи. Лексиката на "говорещите" пациенти е ограничена до минимум думите, използвани в ежедневието (5-15 думи).
Що се отнася до продължителността на живота и общото здравословно състояние на пациентите със синдром на Anghelman, тук цифрите варират средно. В зряла възраст пациентите обикновено се сблъскват със здравословни проблеми като сколиоза и затлъстяване, които при правилния подход към лечението не са животозастрашаващи.