Медицински експерт на статията

Нови публикации
Синдром на Ушер
Последно прегледани: 23.04.2024

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
Синдромът на Ушър е наследствено заболяване, което се проявява под формата на пълна глухота от раждането, както и прогресивна слепота с възрастта. Загубата на зрение е свързана с пигментния ретинит - това е процесът на пигментарна дегенерация на очната ретина. Много хора с синдром на Usher също имат сериозни проблеми с равновесието.
Епидемиология
Поради тези изследвания е лесно да се установи, че около 8% от изследваните глухи деца са болни от синдрома на Usher (тестването е проведено в специални институции за глухи хора). Пигментиран ретинит е наблюдаван при 6-10% от пациентите с вродена глухота, което на свой ред се наблюдава при около 30% от хората с пигментоза на ретинита.
Смята се, че това заболяване се проявява в около 3-10 души от 100 хиляди по целия свят. Тя може да се наблюдава при жените и мъжете. Този синдром засяга около 5-6% от световното население. Около 10% от всички случаи на детска дълбока глухота се дължат на синдрома на Usher I, а също и на тип II.
В САЩ 1 и 2 вида са най-често срещаните типове. Заедно те представляват приблизително 90 до 95 процента от всички случаи на синдром на Usher при деца.
Причини синдром на Usher
Типовете на синдрома на Usher I, II и III носят автозомно рецесивно действие, но тип IV се счита за нарушение на хромозомата X. Причините за този синдром на слепота, както и глухота, не са достатъчно проучени. Предполага се, че хората с това заболяване са свръхчувствителни към компоненти, които могат да увредят структурата на ДНК. С това заболяване могат да се свържат и нарушения на имунната система, но в този случай няма точна картина на такъв процес.
През 1989 г. Пациентите с болест тип II са диагностицирани за първи път с хромозомни аномалии, които впоследствие могат да доведат до начин за изолиране на гени, които задействат развитието на синдрома. Освен това, също така ще бъде възможно да се идентифицират тези гени от техните носители и да се разработят специални генетични тестове за пренатали.
 [8]
[8]
Рискови фактори
Наследяването на синдрома се случва в случай, когато и двамата родители са болни, т.е. Наследството е рецесивен тип. Дете може да наследи заболяване, в случай че родителите му са носители на гена. Ако и двамата бъдещи родители имат този ген, вероятността да има бебе с този синдром е от 1 до 4. Лице, което има само един ген от синдрома, се счита за носител, но самият той няма симптоми на разстройството. Днес все още не е възможно да се определи дали дадено лице има ген за тази болест.
Ако детето се роди на родители, единият от които няма такъв ген, тогава вероятността той да наследи синдрома е много малък, но той ще бъде недвусмислен носител.
Симптоми синдром на Usher
Симптомите на синдрома на Usher са загуба на слуха и освен това патологичното натрупване на пигментирани клетки в очните структури. Освен това, пациентът развива дегенерация на ретината на очите поради това, което започва влошаване на зрението и последвалата загуба в най-тежкия случай.
Сензорната загуба на слуха е лека или пълна и обикновено не напредва от раждането. Но пигментозата на ретинита може да започне да се развива в детството или по-късно. Резултатите от проучването показват, че остротата на централното зрение може да продължи дълго време, дори когато периферното зрение се влоши (това състояние се нарича "виждане на тунела").
Това са основните прояви на болестта, които понякога могат да бъдат допълнени от други разстройства - като психоза и други психични разстройства, проблеми с вътрешното ухо и / или катаракта.
Форми
По време на изследването бяха идентифицирани 3 вида болести, а също и 4 форми - доста редки.
Типът на заболяването се характеризира с вродена пълна глухота, както и с нарушение на равновесието. Често тези деца започват да ходят едва на 1,5 години. Влошаването на зрението обикновено започва с 10 години, а окончателното развитие на състоянието на нощната слепота започва с 20 години. При деца с такъв тип заболяване може да се развие прогресивно влошаване на периферното зрение.
При заболяване от тип II се наблюдава умерена или вродена глухота. Често в този случай не се наблюдава влошаване с частична глухота. Пигментният ретинит започва да се развива в края на юношеския период или след 20 години. Развитието на нощната слепота обикновено започва на 29-31 години. Нарушенията на зрителната острота в случай на патология тип II основно се развиват малко по-бавно, отколкото при тип I.
Тип III заболявания, характеризиращи се с прогресивна загуба на слуха, обикновено започва по време на пубертета, както и постепенно появата на същия период (малко по-късно от глухота) ретинит пигментоза, че може да стане фактор в развитието на прогресивна слепота.
Проявите на четвъртия тип патология се проявяват главно при мъжете. В този случай има и прогресивни нарушения и загуба на слуха и зрението. Тази форма е много рядка и обикновено има характер на Х-хромозома.
Диагностика синдром на Usher
Диагнозата на синдрома на Usher се прави на базата на комбинацията от пациент с внезапна глухота и прогресивна загуба на зрение.
Анализи
За откриване на мутация може да бъде определен специален генетичен тест.
Намерени са 11 генетични локуса, които могат да причинят развитието на синдрома на Ушер и са идентифицирали девет гена, които са точно причината за разстройството:
- Тип 1: MY07A, USH1C, Cdh23, Pcdh15, SANS.
- Тип 2: ush2a, VLGR1, WHRN.
- Тип 3 синдром на Ushira: USH3A.
NIDCD учени, заедно с колеги от университета в Ню Йорк и Израел са идентифицирали мутация, наречена R245X Pcdh15 ген, който е голям процент от тип 1 синдром Ъшър сред еврейското население.
За да научите за лабораториите, които провеждат клинични изпитвания, посетете https://www.genetests.org и потърсете лаборатория за лабораторни изследвания, като напишете термина "синдром на Usher".
За да научите за текущи клинични изпитвания, които включват генетични тестове синдром Ъшър, посетете уеб сайта и да влезе търсенето https://www.clinicaltrials.gov "Ъшър синдром" или "Ъшър синдром генетични тестове."
[25], [26], [27], [28], [29], [30]
Инструментална диагностика
Има няколко метода за инструментална диагностика:
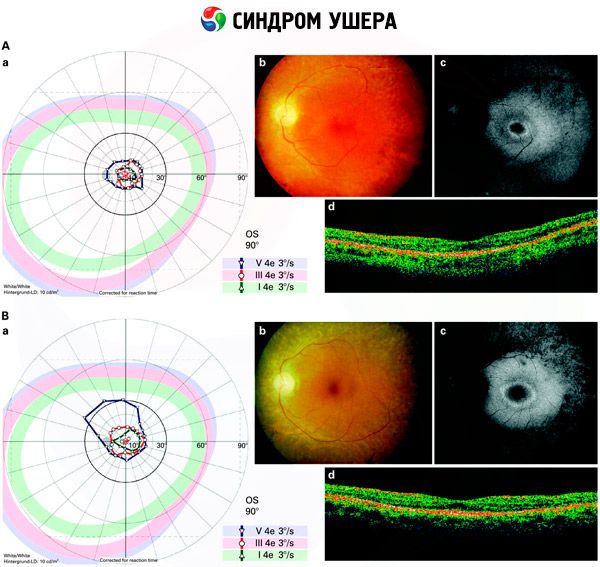
- Изследване на фундуса за идентифициране на присъствието на ретини върху ретината, както и за стесняване на съдовете на ретината;
- Електроретинограма, която ви позволява да идентифицирате първоначалните дегенеративни аномалии в очната ретина. Показва изчезването на електро-рентгенографски пътеки;
- Електронсиграмограмата (ENG) измерва принудителните движения на очите, което може да покаже наличието на дисбаланс
- Аудиометрия, която определя наличието на глухота и степента на нейната тежест.
Диференциална диагноза
Синдромът на Usher трябва да бъде диференциран с някои подобни аномалии.
Синдромът на Халгрен, при който се наблюдава вродена слухова загуба, както и прогресивна загуба на зрение (също има катаракта и нистагъм). Сред допълнителните симптоми на заболяването: атаксия, психомоторни разстройства, психоза и умствено изоставане.
Синдром на Alstrom, който е наследствено заболяване, в което възниква дегенерация на ретината, в резултат на което се губи централно зрение. Този синдром е свързан с проблема за детското затлъстяване. В този случай захарният диабет и загубата на слуха започват да се развиват след 10 години.
Рубелите при бременна жена през първия триместър могат да предизвикат различни аномалии в развитието на детето. Сред последиците от тази аномалия е загуба на слуха, както и (или) проблеми със зрението и освен това различни дефекти в развитието.
Към кого да се свържете?
Лечение синдром на Usher
За да се излекува синдромът на Ушър е невъзможно. Следователно, терапията в този случай е главно да забави процеса на падане на зрението и да компенсира загубата на слуха. Възможните третирания включват:
- Използването на витамин А група (някои офталмолози вярват, че високи дози витамин А палмитат могат да забавят, но не и да спрат, прогресията на пигментния ретинит);
- Имплантиране на специални електронни устройства в ушната мида на пациента (слухов апарат, кохлеарни импланти.
Офталмолозите препоръчват на повечето възрастни пациенти с напреднали форми на пигмент за ретинит да приемат 15 000 IU (международни единици) витамин А дневно под формата на палмитат под наблюдение. Тъй като хората с синдром на Usher тип 1 не са участвали в проучването, високи дози витамин А не се препоръчват за тази група пациенти. Хората, които обмислят приема на витамин А, трябва да обсъдят тази възможност с лекаря си. Други препоръки относно тази опция за лечение включват:
- Променете вашата диета с включването на храни с високо съдържание на витамин А.
- Жените, които планират бременност, трябва да спрат да приемат високи дози витамин А три месеца преди планираното зачеване поради повишения риск от вродени дефекти.
- Жените, които са бременни, трябва да спрат да приемат високи дози витамин А поради повишен риск от вродени дефекти.
Също така е важно детето да се адаптира към социалния живот. Това изисква помощта на педагози-дефектолози, както и психолози. В случай, че пациентът е започнал постепенно понижаване на визията, човек трябва да го научи да използва езика на жестовете.
Прогноза
Синдромът на Ушър има неблагоприятна прогноза. Зрителното поле и неговата сериозност започват да се влошават в период от 20-30 години при повечето пациенти с това заболяване от всякакъв вид. В някои случаи става въпрос за пълна двустранна загуба на зрение. Глухотата, която винаги се наблюдава и глупост, много бързо се развива до пълна двустранна загуба на слуха.