Медицински експерт на статията

Нови публикации
Синдром на Апер
Последно прегледани: 04.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.

 [
[ Причини Синдром на Апер
Развитието на синдрома се провокира от нарушение в структурата на ген, разположен на хромозома 10. Този ген е отговорен за процеса на разделяне на пръстите и освен това за навременното затваряне на черепните шевове.
Освен това, инфекциозни заболявания, прекарани по време на бременност (като рубеола или туберкулоза, сифилис или грип, както и менингит), и излагане на майката на рентгенови лъчи се считат за причини за патологията. Най-често такъв синдром се открива при деца, родени от възрастни родители.
Патогенеза
Синдромът на Аперт е наследствен. Типът му е автозомно доминантен (т.е. в семейство, в което един от бъдещите родители е болен, вероятността да се роди бебе с това заболяване е 50-100%).
Уникална мутация в рецептора 2 на растежния фактор на фибробластите (FGFR2) води до увеличен брой прогениторни клетки, които се развиват по остеогенния път. Това в крайна сметка води до повишено образуване на субпериостална костна матрица и преждевременна осификация на черепните шевове по време на феталното развитие. Редът и скоростта на топене на шевовете определят степента на деформация и увреждане. След като шевният материал заздравее, растежът на други тъкани, перпендикулярни на този шев, е ограничен и срасналите кости действат като единна костна конструкция.
Първото генетично доказателство, че синдактилията при синдрома на Аперт се дължи на дефект в рецептора на кератиноцитния растежен фактор (KGFR), е наблюдението на корелация между експресията на KGFR във фибробластите и тежестта на синдактилията.
Амблиопията и страбизмът са по-чести при пациенти с мутация FGFR2 Ser252Trp, а атрофията на зрителния диск е по-честа при пациенти с мутация FGFR2 Pro253Arg. Пациентите с мутации FGR2 Ser252Trp имат значително по-висока честота на зрителни увреждания в сравнение с пациентите с мутация FGFR2 Pro253Arg.
Симптоми Синдром на Апер
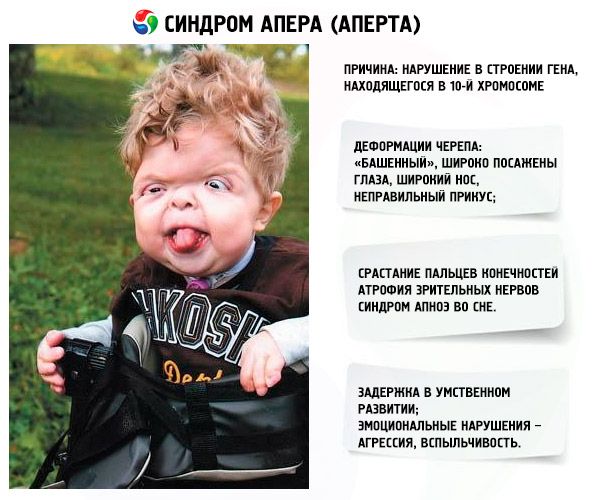
Някои прояви на заболяването са ясно забележими още при раждането на бебето, тъй като се развиват в утробата на майката. Сред основните симптоми на синдрома са:
- Черепът е деформиран - той се разтяга на височина, придобивайки вид на „кула“; освен това очите са широко разположени и леко изпъкнали (защото размерът на очните кухини намалява); носът се разширява и се образува неправилна захапка (горните зъби стърчат прекомерно);
- Пръстите на крайниците са напълно сраснали (главно безименният пръст, както и средният и показалеца) и изглеждат като кожна мембрана или пълно сливане на кости; освен това могат да растат допълнителни пръсти;
- Умствена изостаналост (не се среща при всички);
- Зрителният нерв атрофира, в резултат на което зрителната острота намалява (в някои случаи се развива пълна загуба на зрение);
- Повишеното вътречерепно налягане, което възниква в резултат на преждевременно затваряне на черепните конци, се проявява под формата на главоболие и повръщане с гадене;
- Тъй като горната челюст остава недоразвита, възникват проблеми с дишането;
- Синдромът на сънна апнея е често срещано явление.
- Емоционални прояви – агресия, липса на сдържаност, силна раздразнителност.
Усложнения и последствия
Повечето пациенти имат известна степен на обструкция на горните дихателни пътища в ранна детска възраст. Горните дихателни пътища са слабо проходими поради намаления размер на назофаринкса и хоаните, а долните дихателни пътища могат да причинят ранна смърт поради аномалии на трахеалния хрущял.
Диагностика Синдром на Апер
За да се постави диагноза, са необходими следните стъпки:
- Лекарят трябва да анализира оплакванията на пациента, както и медицинската история. Необходимо е да се установи дали е имало случаи на подобна патология в семейството;
- Неврологичен преглед за оценка на формата на черепа и интелектуалното развитие на пациента (специални въпросници, както и интервю);
- Изследване на фундуса за откриване на наличието на симптоми на повишено вътречерепно налягане (подуване на зрителния диск, както и замъгляване на краищата му);
- За да се оцени състоянието на черепа, се извършва рентгенова снимка;
- Компютърно и магнитно-резонансно изобразяване на главата за изследване на структурата на мозъка и черепа слой по слой, за определяне на наличието на симптоми на преждевременно срастване на черепните шевове, а освен това и хидроцефалия (поради повишено вътречерепно налягане се натрупва излишна цереброспинална течност (това е цереброспиналната течност, която насърчава метаболитния процес, както и храненето на мозъка));
- Рентгенова снимка на краката и ръцете, за да се определи причината за сливането на пръстите (това е важно за планирането на последваща хирургическа интервенция);
- Може да бъде предписана консултация с медицински генетик и неврохирург.
Тестове
Провежда се генетичен анализ на често срещани мутации, срещащи се в гена от типа FGFR2.
Диференциална диагноза
Този синдром трябва да се диференцира от други генетични патологии, при които се наблюдава краниосиностоза. Това са заболявания като синдромите на Pfeiffer, Crouzon, Saethre-Chotzen и Carpenter. Молекулярно-генетичните методи за изследване се използват за изключване на тези аномалии.
Към кого да се свържете?
Лечение Синдром на Апер
Хирургията се счита за единственото ефективно лечение за синдрома на Аперт – тя помага за коригиране на индивидуални физически дефекти, а също така коригира умствената изостаналост.
По време на тази процедура коронарният шев се затваря, за да се предотврати евентуално мозъчно увреждане. Най-често използваната техника е краниофациална дистракция, която включва градуирана тракция на черепа. Ортодонтска и/или ортогнатична хирургия се извършва за отстраняване на отделни лицеви дефекти.
Освен това, пациентите се подлагат на хирургична корекция на сливането на пръстите.
Референции
Медицинска генетика. Национално ръководство. Под редакцията на Ginter EK, Puzyrev VP GEOTAR-Media. 2022 г