Медицински експерт на статията

Нови публикации
Полимиозит и дерматомиозит: причини, симптоми, диагноза, лечение
Последно прегледани: 05.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.
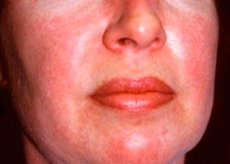
Полимиозитът и дерматомиозитът са редки системни ревматични заболявания, характеризиращи се с възпалителни и дегенеративни промени в мускулите (полимиозит) или мускулите и кожата (дерматомиозит). Най-специфичната кожна проява е хелиотропен обрив.
Мускулното засягане е симетрично и включва слабост, известна болезненост и последваща атрофия на мускулите на проксималния тазов пояс. Усложненията могат да включват висцерално засягане и злокачествено заболяване. Диагнозата се основава на клиничната картина и оценка на мускулната дисфункция чрез измерване на ензимните нива, извършване на ЯМР, електромиография и мускулна биопсия. Лечението включва глюкокортикоиди, понякога в комбинация с имуносупресори или интравенозни имуноглобулини.
Жените боледуват два пъти по-често от мъжете. Заболяването може да се появи на всяка възраст, но най-често се открива в диапазона от 40 до 60 години; при деца - от 5 до 15 години.
 [
[ Какво причинява дерматомиозит и полимиозит?
Смята се, че причината за заболяването е автоимунна реакция към мускулната тъкан при генетично предразположени индивиди. Заболяването е по-често срещано при наличие на обременена фамилна анамнеза и носители на определени HLA антигени (DR3, DR52, DR56). Възможни тригери са вирусен миозит и злокачествени неоплазми. Има съобщения за откриване на структури, подобни на пикорнавирусите, в мускулните клетки; освен това вирусите могат да индуцират подобни заболявания при животни. Асоциацията на злокачествените тумори с дерматомиозит (много по-рядко, отколкото с полимиозит) предполага, че туморният растеж също може да бъде спусък за развитието на заболяването в резултат на започване на автоимунни реакции към общи антигени на тумора и мускулната тъкан.
В стените на кръвоносните съдове на скелетните мускули се откриват отлагания на IgM, IgG и третия компонент на комплемента; това е особено характерно за дерматомиозит при деца. Пациентите с полимиозит могат да развият и други автоимунни процеси.
Патофизиология на дерматомиозит и полимиозит
Патологичните промени включват клетъчно увреждане и атрофия на фона на възпаление с различна степен на тежест. Мускулите на горните и долните крайници, както и лицето, са увредени в по-малка степен от другите скелетни мускули. Увреждането на висцералните мускули на фаринкса и горната част на хранопровода, по-рядко на сърцето, стомаха или червата, може да доведе до нарушена функция на гореспоменатите органи. Високите концентрации на миоглобин, дължащи се на рабдомиолиза, могат да причинят увреждане на бъбреците. Възможно е да се появят и възпалителни промени в ставите и белите дробове, особено при пациенти, които имат антисинтетазни антитела.
Симптоми на дерматомиозит и полимиозит
Началото на полимиозита може да бъде остро (особено при деца) или подостро (по-често при възрастни). Острата вирусна инфекция понякога предшества или е спусък за проявата на заболяването, като най-честите прояви са слабост на проксималните мускули или кожни обриви. Болката е изразена в по-малка степен, отколкото слабостта. Могат да се развият полиартралгия, феномен на Рейно, дисфагия, белодробни нарушения, общи симптоми (повишена телесна температура, намалено телесно тегло, слабост). Феноменът на Рейно често се среща при пациенти със съпътстващи заболявания на съединителната тъкан.
Мускулната слабост може да прогресира в продължение на няколко седмици или месеци. Въпреки това, за клинично проявление на мускулна слабост, трябва да бъдат засегнати поне 50% от мускулните влакна (по този начин наличието на мускулна слабост показва прогресия на миозит). Пациентите могат да изпитват затруднения при повдигане на ръцете над нивото на раменете, при изкачване на стълби или при ставане от седнало положение. Поради силна слабост на мускулите на таза и раменния пояс, пациентите могат да бъдат ограничени до инвалидна количка или легло. Ако са засегнати флексорите на врата, става невъзможно да се повдигне главата от възглавницата. Засягането на мускулите на фаринкса и горния хранопровод води до нарушения на преглъщането и регургитация. Мускулите на долните, горните крайници и лицето обикновено не са засегнати. Въпреки това, могат да се развият контрактури на крайниците.
Кожните обриви, наблюдавани при дерматомиозит, обикновено са с тъмен цвят и еритематозни. Характерен е и периорбитален оток с лилав цвят (хелиотропен обрив). Кожните обриви могат да бъдат леко повдигнати над нивото на кожата и да бъдат гладки или люспести; локализацията на обривите е челото, шията, раменете, гърдите, гърба, предмишниците, долните части на пищялите, веждите, областта на коленете, медиалните малеоли, гръбните повърхности на интерфалангеалните и метакарпофалангеалните стави, от страничната страна (симптом на Готрон). Възможна е хиперемия на основата или периферията на ноктите. Десквамативен дерматит, придружен от пукнатини, може да се развие по кожата на страничната повърхност на пръстите. Първичните кожни лезии често отшумяват без последствия, но могат да доведат до вторични промени като тъмна пигментация, атрофия, белези или витилиго. Могат да се развият подкожни калцификати, особено при деца.
Приблизително 30% от пациентите развиват полиартралгия или полиартрит, често съпроводени с оток и ставен излив. Тежестта на ставните прояви обаче е малка. Те се появяват по-често, когато при пациентите се открият антитела срещу Jo-1 или други синтетази.
Засягането на вътрешни органи (с изключение на фаринкса и горната част на хранопровода) е по-рядко срещано при полимиозит, отколкото при други ревматични заболявания (особено системен лупус езофагит и системна склероза). Рядко, особено при антисинтетазен синдром, заболяването се проявява като интерстициален пневмонит (под формата на диспнея и кашлица). Могат да се развият сърдечни аритмии и нарушения на проводимостта, но те обикновено са асимптоматични. Стомашно-чревните прояви са по-чести при деца, които също имат васкулит, и могат да включват повръщане с кръв, мелена и чревна перфорация.
Къде боли?
Класификация на полимиозита
Има 5 вида полимиозит.
- Първичният идиопатичен полимиозит, който може да се появи във всяка възраст, не засяга кожата.
- Първичният идиопатичен дерматомиозит е подобен на първичния идиопатичен полимиозит, но засяга кожата.
- Полимиозит и дерматомиозит, свързани със злокачествени новообразувания, могат да се появят при пациенти на всяка възраст; развитието им се наблюдава най-често при пациенти в напреднала възраст, както и при пациенти с други заболявания на съединителната тъкан. Развитието на злокачествени новообразувания може да се наблюдава както в рамките на 2 години преди, така и в рамките на 2 години след началото на миозита.
- Детският полимиозит или дерматомиозит е свързан със системен васкулит.
- Полимиозит и дерматомиозит могат да се появят и при пациенти с други заболявания на съединителната тъкан, най-често прогресивна системна склероза, смесено заболяване на съединителната тъкан и системен лупус еритематозус (СЛЕ).
Включването на миозита на мускулите на торса в групата на полимиозитите е неправилно, тъй като последното е отделно заболяване, характеризиращо се с клинични прояви, подобни на тези на хроничния идиопатичен полимиозит. То обаче се развива в напреднала възраст, често засяга мускулите на дисталните части на тялото (например горните и долните крайници), има по-голяма продължителност, реагира по-слабо на лечение и се характеризира с типична хистологична картина.
Диагностика на дерматомиозит и полимиозит
Полимиозит трябва да се подозира при пациенти с оплаквания от слабост на проксималните мускули, със или без чувствителност. Оценка за дерматомиозит е необходима при пациенти с оплаквания от обрив, наподобяващ хелиотроп или симптом на Готрон, както и при пациенти с прояви на полимиозит, комбинирани с кожни лезии, съответстващи на дерматомиозит. Клиничните прояви на полимиозит и дерматомиозит могат да наподобяват тези на системна склероза или, по-рядко, на системен лупус еритематозус (СЛЕ) или васкулит. Сигурността на диагнозата се повишава чрез отговаряне на възможно най-много от следните пет критерия:
- слабост на проксималните мускули;
- характерни кожни обриви;
- повишена активност на ензимите в мускулната тъкан (креатин киназа или, при липса на повишаване на нейната активност, аминотрансферази или алдолаза);
- характерни промени в миографията или ЯМР;
- характерни хистологични промени в биопсия на мускулна тъкан (абсолютен критерий).
Мускулната биопсия може да изключи някои клинично подобни състояния, като миозит на мускулите на торса и рабдомиолиза, дължаща се на вирусна инфекция. Промените, разкрити чрез хистологично изследване, могат да варират, но хроничното възпаление, огнищата на мускулна дегенерация и регенерация са типични. Необходима е точна диагноза (обикновено чрез хистологична проверка) преди започване на потенциално токсично лечение. Ядрено-магнитният резонанс (ЯМР) може да открие огнища на оток и възпаление в мускулите, последвано от тяхната целенасочена биопсия.
Лабораторните изследвания могат да потвърдят или, напротив, да елиминират съмнението за наличие на заболяването, а също така са полезни при оценката на неговата тежест, възможността за комбинация с друга подобна патология и диагностицирането на усложнения. Въпреки факта, че при някои пациенти се откриват антинуклеарни антитела, това явление е по-типично за други заболявания на съединителната тъкан. Около 60% от пациентите имат антитела към антигена на ядрата (PM-1) или цели тимусни клетки и Jo-1. Ролята на автоантителата в патогенезата на заболяването остава неясна, въпреки че е известно, че антителата към Jo-1 са специфичен маркер на антисинтетазен синдром, включително фиброзиращ алвеолит, белодробна фиброза, артрит и феномен на Рейно.
Периодичната оценка на активността на креатин киназата е полезна за мониторинг на лечението. Въпреки това, при пациенти с тежка мускулна атрофия, ензимната активност може да е нормална, въпреки наличието на хроничен активен миозит. ЯМР, мускулна биопсия или повишена активност на креатин киназата често са полезни за разграничаване между рецидив на полимиозит и миопатия, индуцирана от глюкокортикоиди.
Тъй като много пациенти имат недиагностицирани злокачествени заболявания, някои автори препоръчват скрининг на всички възрастни с дерматомиозит и тези с полимиозит над 60-годишна възраст, като се използва следният график: физикален преглед, включително прегледи на гърдите, таза и ректума (включително изследване на изпражненията за окултна кръв); пълна кръвна картина; кръвна химия; мамография; изследване на карциноембрионен антиген; анализ на урината; рентгенография на гръдния кош. Някои автори поставят под въпрос необходимостта от такъв скрининг при по-млади пациенти, които нямат клинични данни за злокачествено заболяване.
Какво трябва да проучим?
Как да проучим?
Лечение на дерматомиозит и полимиозит
Докато възпалението не се облекчи, физическата активност трябва да бъде ограничена. Глюкокортикоидите са лекарства от първа линия. В острия стадий на заболяването на възрастни пациенти трябва да се предписва преднизолон (перорално) в доза от 40 до 60 mg на ден. Редовното определяне на креатин киназната активност е ранен индикатор за ефективност: при повечето пациенти се наблюдава намаление или нормализиране в рамките на 6 до 12 седмици след повишаване на мускулната сила. След нормализиране на ензимната активност дозата на преднизолон се намалява: първо с приблизително 2,5 mg на ден в продължение на една седмица, след това по-бързо; ако повишената мускулна ензимна активност рецидивира, хормоналната доза се увеличава отново. Възстановените пациенти могат да се справят без глюкокортикоиди, но най-често възрастните пациенти се нуждаят от дългосрочна глюкокортикоидна терапия (10-15 mg преднизолон на ден). Началната доза преднизолон за деца е 30-60 mg/m2 веднъж дневно. При наличие на ремисия за > 1 година при деца, глюкокортикоидната терапия може да бъде прекратена.
В някои случаи пациентите, получаващи високи дози глюкокортикоиди, изпитват внезапно повишаване на мускулната слабост, което може да е свързано с развитието на глюкокортикоидна миопатия.
В случай на неадекватен отговор на лечение с глюкокортикоиди, както и в случай на развитие на глюкокортикоидна миопатия или други усложнения, изискващи намаляване на дозата или прекратяване на приема на преднизолон, трябва да се използват имуносупресори (метотрексат, циклофосфамид, азатиоприн, циклоспорин). Някои пациенти могат да получават само метотрексат (обикновено в дози, превишаващи използваните за лечение на ревматоиден артрит) повече от 5 години. Интравенозните имуноглобулини могат да бъдат ефективни при пациенти, рефрактерни на лекарствена терапия, но употребата им увеличава разходите за лечение.
Миозитът, свързан с първични и метастатични тумори, както и миозитът на мускулите на торса, обикновено са по-резистентни към глюкокортикоидна терапия. Ремисия на миозит, свързан със злокачествени тумори, е възможна след отстраняване на тумора.
Каква е прогнозата за дерматомиозит и полимиозит?
Дългосрочна ремисия (и дори клинично възстановяване) над 5 години се наблюдава при повече от половината от лекуваните пациенти; при децата тази цифра е по-висока. Рецидив обаче може да възникне по всяко време. Общата петгодишна преживяемост е 75%, като е по-висока при децата. Причините за смърт при възрастни са тежка и прогресираща мускулна слабост, дисфагия, намалено хранене, аспирационна пневмония или дихателна недостатъчност, дължаща се на белодробни инфекции. Полимиозитът е по-тежък и резистентен на лечение, ако има увреждане на сърцето и белите дробове. Смърт при децата може да настъпи поради чревен васкулит. Общата прогноза на заболяването се определя и от наличието на злокачествени новообразувания.