Медицински експерт на статията

Нови публикации
Синдром на Пиер Робен
Последно прегледани: 04.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.

Синдромът на Пиер-Робин, известен в медицината още като аномалия на Робин, е вродена патология на развитието на челюстната част на лицето. Заболяването получава името си в чест на френския зъболекар П. Робин, който пръв описва всички негови признаци. Ланелонг и Менар описват за първи път синдрома на Пиер-Робин през 1891 г. в своя доклад за двама пациенти с микрогнатия, цепнато небце и ретроглосоптоза. През 1926 г. Пиер-Робин публикува случай на заболяването при кърмаче с признаци на класическия синдром. До 1974 г. триадата от признаци е била известна като синдром на Робин-Пиер. Сега обаче този синдром се използва за описание на малформации с едновременно наличие на множество аномалии.
Епидемиология
Това е хетерогенен вроден дефект, който има честота 1 на 8500 живородени деца. Съотношението мъже и жени е 1:1, с изключение на Х-свързаната форма.
Сред тези пациенти 50% от бебетата имат непълна цепка на мекото небце, останалите се раждат със сводесто и необичайно високо небце, но без цепка.
Причини Синдром на Пиер Робен
Разглежда се възможността за автозомно-рецесивно унаследяване на заболяването. Съществуват два вида синдром в зависимост от етиологията: изолиран и генетично обусловен. Изолираният тип се развива поради компресия на долната част на челюстта по време на ембрионалното развитие. Компресията може да се развие поради:
- Наличието на локализирани уплътнения в матката (кисти, белези, тумори).
- Многоплодна бременност.
Също така, развитието на челюстта при плода може да бъде нарушено от:
- Вирусни инфекции, които бъдещата майка е претърпяла по време на бременност.
- Невротрофични разстройства.
- Недостатъчно количество фолиева киселина в тялото на бременна жена.
Патогенеза
Синдромът на Пиер Робин се причинява от ембрионални нарушения, причинени от голямо разнообразие от патологии в пренаталния период.
Съществуват три патофизиологични теории, които могат да обяснят появата на синдрома на Пиер Робин.
Механична теория: Тази теория е най-вероятната. Недоразвитието на мандибуларния апарат настъпва между 7-та и 11-та седмица от бременността. Високото положение на езика в устната кухина води до образуване на цепки в небцето, поради които кухата вена не се затваря. Тази теория обяснява класическата обърната U-образна цепка и липсата на свързаната с нея цепка на устната. Олигохидрамнионът може да играе роля в етиологията, тъй като липсата на околоплодна течност може да доведе до деформация на брадичката и последващо компресиране на езика между кухата вена.
Неврологична теория: Забавяне в неврологичното развитие е наблюдавано с електромиография на мускулите на езика и фарингеалните колони, както и на вкуса, поради забавяне на проводимостта в хипоглосалния нерв.
Теория за дисневрорегулацията на ромбенцефалона: Тази теория се основава на нарушаването на развитието на ромбенцефалона по време на онтогенезата.
Недостатъчното развитие на долната част на челюстта на детето води до значително намаляване на устната кухина. Това от своя страна причинява така наречената псевдомакроглосия, т.е. езикът е изместен към задната част на фарингеалната стена. Тази патология води до развитие на обструкция на дихателните пътища.
Докато бебето плаче или се движи, дихателните пътища остават свободни, но веднага щом бебето заспи, запушването се появява отново.
Поради дихателни нарушения процесът на хранене на бебето е много труден. По това време почти винаги се наблюдава запушване на дихателните пътища. Ако не се приложи медицинска корекция, подобна патология може да доведе до тежко изтощение на цялото тяло и дори до смърт.
Симптоми Синдром на Пиер Робен
Заболяването се характеризира с три основни симптома:
- Долна микрогнатия (недоразвитие на долната челюст, среща се в 91,7% от случаите на заболяването). Характеризира се с прибиране на долната зъбна дъга с 10-12 мм зад горната дъга. Долната челюст е с малко тяло, тъп ъгъл. Детето постига нормално развитие приблизително на 5-6-годишна възраст.
- Глосоптоза (прибиране на езика поради недостатъчното му развитие, наблюдавано в 70-85% от случаите).
- Макроглосията и анкилоглосията са сравнително редки симптоми, наблюдавани в 10-15% от случаите.
- В небето се появява пукнатина.
- Брадипнея и диспнея.
- Лека цианоза.
- Асфиксия, която най-често се случва по време на опити за хранене на бебето.
- Преглъщането е невъзможно или много трудно.
- Усещане за повръщане.
- Аурикуларни аномалии в 75% от случаите.
- Кондуктивна загуба на слуха се среща при 60% от пациентите, докато атрезия на външния слухов канал се среща само при 5% от пациентите, недостатъчна пневматизация на мастоидната кухина на темпоралната кост.
- Аномалии на вътрешното ухо (аплазия на страничните полукръгли канали, голям вестибуларен акведукт, загуба на кохлеарни власинкови клетки).
- Носните малформации са рядко срещани и се състоят главно от аномалии на корена на носа.
- Зъбните малформации се срещат в 30% от случаите. Ларингомалация и велофарингеална недостатъчност се срещат при приблизително 10-15% от пациентите със синдром на Пиер Робен.
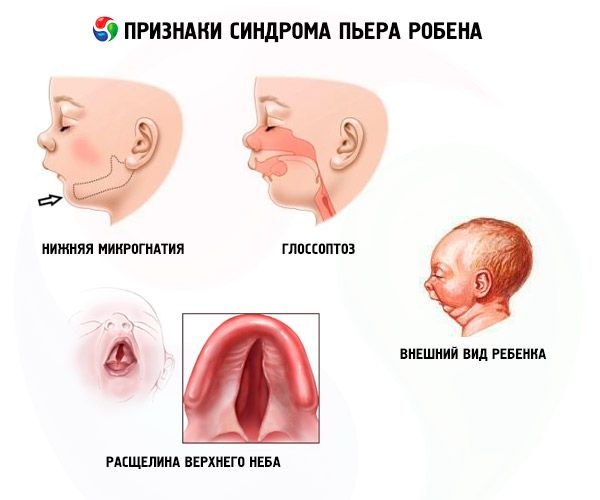
Системни характеристики на синдрома на Пиер Робин
Системни аномалии в развитието са описани в 10-85% от регистрираните случаи.
Очни аномалии се срещат при 10-30% от пациентите. Те могат да включват: хиперопия, миопия, астигматизъм, склероза на роговицата и стеноза назолакрималния канал.
Сърдечно-съдови патологии: доброкачествени сърдечни шумове, стеноза на белодробната артерия, открит дуктус артериозус, овален прозорец, дефект на междупредсърдната преграда и белодробна хипертония. Тяхната честота варира от 5 до 58%.
Аномалии, свързани с опорно-двигателния апарат (70-80% от случаите): синдактилия, диспластични фаланги, полидактилия, клинодактилия, хипермобилност на ставите и олигодактилия на горните крайници. Аномалии на долните крайници: аномалии на стъпалото (клишостъп, метатарзална аддукция), малформации на бедрената кост (валгусен или варусен таз, къси бедрени кости), аномалии на тазобедрената става (вродена луксация, контрактури), аномалии на колянните стави (GENU VALGUS, синхондроза). Малформации на гръбначния стълб: сколиоза, кифоза, лордоза, вертебрална дисплазия, агенезия на сакрума и опашния синус.
Патология на централната нервна система: епилепсия, забавяне на развитието на нервната система, хидроцефалия. Честотата на дефектите на ЦНС е около 50%.
Генитоуринарни аномалии: неспуснати тестиси (25%), хидронефроза (15%) и хидроцеле (10%).
Свързани синдроми и състояния: синдром на Стиклър, синдром на тризомия 11q, тризомия 18, синдром на делеция 4q, ревматоидна артропатия, хипохондроплазия, синдром на Мьобиус.
Етапи
Има три степени на тежест на заболяването, които зависят от състоянието на дихателните пътища на детето:
- Лека форма - има леки проблеми с храненето, но дишането почти не е затруднено. Лечението се провежда амбулаторно.
- Умерено – дишането е умерено затруднено, храненето на детето е умерено затруднено. Лечението се провежда в болница.
- Тежко – дишането е много затруднено, детето не може да се храни нормално. Необходимо е използването на специални устройства (интраназална сонда).
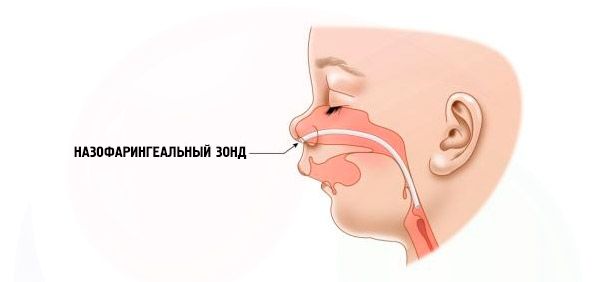
Усложнения и последствия
Комбинацията от микрогнатия и глосоптоза може да доведе до тежки респираторни усложнения и проблеми по време на хранене на детето.
Синдромът на Пиер Робин причинява следните усложнения:
- Стридозно дишане поради обструкция на дихателните пътища. Ларингомалация или дори сънна асфиксия.
- Психомоторното развитие на детето изостава значително от това на връстниците му.
- Физическото развитие също изостава.
- Речта на пациентите е нарушена.
- Чести ушни инфекции, които стават хронични и водят до увреждане на слуха.
- Синдром на обструктивна сънна апнея, при който смъртта по време на сън варира в 14-91% от случаите.
- Проблеми със зъбите.
Диагностика Синдром на Пиер Робен
Диагнозата на синдрома на Пиер Робин не е трудна. Тя се основава на клинични прояви. За да се изключат други патологии, е много важно да се консултирате с генетик.
Децата с вродена аномалия на Робин имат проблеми с дишането от раждането си, поради постоянното потъване на езика назад. Бебето е неспокойно, кожата му е синкава, хрипове излизат от гърдите при вдишване. По време на хранене може да се появи задавяне. Диагнозата може да се постави и по необичайния външен вид на детето - „птиче лице“. Често пациентите развиват и други дефекти: късогледство, катаракта, патология на пикочно-половата система, сърдечна патология, аномалии в развитието на гръбначния стълб.
Въз основа на тези клинични прояви, за специалист няма да е трудно да постави правилна диагноза.
Към кого да се свържете?
Лечение Синдром на Пиер Робен
Лечението се провежда веднага след раждането на дете със синдром на Пиер Робин. Ако заболяването е леко, за да се подобри състоянието на пациента, е необходимо постоянно да се държи детето вертикално или да лежи по корем. Главата на бебето трябва да е наклонена към гърдите. По време на хранене не се препоръчва детето да се държи в хоризонтално положение, за да не попадне храна в дихателните пътища.
Ако недоразвитието на долната челюст е доста изразено, се използва хирургична интервенция, за да се приведе прибраният език в нормално физиологично положение. В тежки случаи езикът се издърпва нагоре и се фиксира върху долната устна. В много тежки случаи се налага да се извърши трахеостомия, глосопексия и дистракционна остеогенеза на долната челюст.
Използва се и консервативно лечение.
Лекарства
Фенобарбитал. Сънотворно и успокоително лекарство, има антиконвулсивен ефект. Всяка таблетка съдържа 100 мл фенобарбитал. Дозировката е индивидуална, тъй като зависи от тежестта на заболяването и състоянието на детето. Лекарството е забранено за пациенти с чернодробна недостатъчност, хиперкинеза, анемия, миастения, порфирия, захарен диабет, депресия и непоносимост към компонентите. При прием са възможни следните симптоми: замаяност, астения, халюцинации, агранулоцитоза, гадене, ниско кръвно налягане и алергии.
Клоназепам. Лекарство, предписвано за лечение на епилепсия. Лекарството съдържа активното вещество клоназепам, което е бензодиазепиново производно. Има антиконвулсивно, анксиолитично и мускулно-релаксиращо действие. Дозата се определя от лекуващия лекар, но не трябва да надвишава максималната - 250 мкг на ден. Да не се приема при безсъние, мускулна хипертонус, психомоторна възбуда, панически разстройства. При прием са възможни следните симптоми: летаргия, гадене, дисменорея, главоболие, левкопения, задържане или инконтиненция на урина, алопеция, алергия.
Сибазон. Предлага се под формата на разтвор и ректални таблетки. Активното вещество е бензодиазепиново производно (сибазон). Има седативен, анксиолитичен, антиконвулсивен ефект. Дозировката е индивидуална. Пациенти с хронична хиперкапния, миастения, непоносимост към бензодиазепини са забранени да приемат лекарството. При употреба на лекарството могат да се развият следните симптоми: гадене, запек, главоболие, световъртеж, хълцане, уринарна инконтиненция, алергии.
Кортексинов лиофилизат. Лекарство с ноотропен ефект. Лекарството съдържа комплекс от водоразтворими полипептидни фракции и глицин. Дозировката е индивидуална и се предписва от лекуващия лекар в съответствие със състоянието на пациента. Пациенти с непоносимост към кортексин не трябва да приемат лекарството. Лекарството може да причини алергични реакции.
Физиотерапевтично лечение
Обикновено, при леки стадии на синдрома, се използва позиционна терапия, при която детето се поставя по корем в изправено положение, докато гравитацията не принуди долната челюст да расте правилно.
Хирургично лечение
Хирургичното лечение се използва предимно за коригиране на глосоптозата. Има няколко метода:
- Поддържане на езика със сребърна нишка. Нишката се прекарва през долната част на венеца и долната устна. Методът се нарича Дъглас.
- Метод на Дюамел - дебела сребърна нишка се прекарва през основата на езика на пациента и двете бузи. Да се използва не повече от тридесет дни.
- Ортопедични устройства за удължаване и фиксиране на езика.
- На едногодишна възраст може да се извърши операция за коригиране на цепнато небце.
Прогноза
Прогнозата и протичането на заболяването са тежки. Най-често смъртта настъпва в първите дни от живота при умерен и тежък стадий на заболяването (причината е асфиксия). Също така, рискът от смърт през първата година е доста висок поради множество инфекции.
За пациенти над двегодишна възраст прогнозата е благоприятна.
 [ 36 ]
[ 36 ]