Нови публикации
Нови открития допринасят за по-доброто разбиране на причините за синдрома на Рет
Последно прегледани: 02.07.2025

Цялото съдържание на iLive е медицински прегледано или е проверено, за да се гарантира възможно най-голяма точност.
Имаме строги насоки за снабдяване и само свързваме реномирани медийни сайтове, академични изследователски институции и, когато е възможно, медицински проучвания, които се разглеждат от специалисти. Имайте предвид, че номерата в скоби ([1], [2] и т.н.) са линкове към тези проучвания.
Ако смятате, че някое от съдържанието ни е неточно, остаряло или под съмнение, моля, изберете го и натиснете Ctrl + Enter.

Синдромът на Рет е рядко неврологично разстройство, за което понастоящем няма лек или добро лечение. Той причинява тежки физически и когнитивни симптоми, много от които се припокриват с разстройства от аутистичния спектър.
Синдромът на Рет се причинява от мутации в гена MECP2, който е силно експресиран в мозъка и изглежда играе важна роля за поддържане на здравето на невроните. Генът е разположен на Х хромозомата и синдромът засяга предимно момичетата. За да разработят лечения за синдрома на Рет, изследователите искат да разберат по-добре MECP2 и неговите функции в мозъка.
Изследователи, включително съоснователят на Института Уайтхед Рудолф Йениш, изучават MECP2 от десетилетия, но много основни факти за гена остават неизвестни. Протеинът, кодиран от гена, MECP2, участва в генната регулация; той се свързва с ДНК и влияе върху нивата на експресия на различни други гени или върху количеството протеин, което те произвеждат.
Въпреки това, изследователите не са разполагали с пълен списък на гените, засегнати от MECP2, и не е имало консенсус относно това как MECP2 влияе на тези гени.
Ранните изследвания на MECP2 предполагат, че той е репресор, намалявайки експресията на целевите си гени, но изследвания на Йениш и други вече са показали, че MECP2 действа и като активатор, увеличавайки експресията на своите цели – и че може би е активатор на първо място. Неизвестен е и механизмът на действие на MECP2 или какво точно прави протеинът, за да предизвика промени в генната експресия.
Ограниченията в технологиите са попречили на изследователите да получат яснота по тези въпроси. Но Яниш, постдокторантът от неговата лаборатория Йи Лиу и бившият член на лабораторията на Яниш Антъни Фламие, сега асистент в изследователския център CHU Sainte-Justine в Университета на Монреал, са използвали авангардни техники, за да отговорят на тези оставащи въпроси относно MECP2 и да получат нови прозрения за неговата роля в здравето и заболяванията на мозъка.
Резултатите им бяха публикувани в списанието Neuron, а изследователите създадоха и онлайн хранилище на своите MECP2 данни, портала MECP2-NeuroAtlas, като ресурс за други изследователи.
„Мисля, че тази статия ще промени коренно разбирането на хората за това как MECP2 причинява синдрома на Рет. Имаме напълно ново разбиране за механизма и това може да осигури нови възможности за разработване на лечения за заболяването“, казва Яниш, който е и професор по биология в MIT.
По-задълбочено разбиране на MECP2 в мозъка
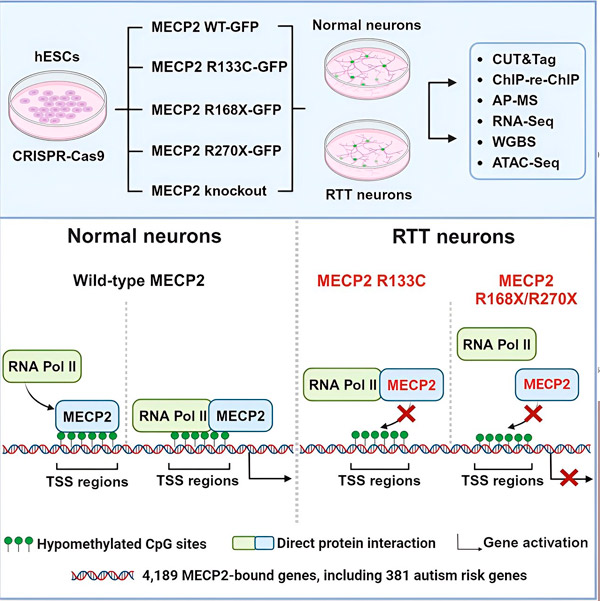
Изследователите първо създадоха подробна карта на местата, където MECP2 се свързва в човешки невронни генни последователности, или в самите гени, или в регулаторни области на ДНК близо до тях. Те използваха подход, наречен CUT&Tag, който може да определи точно взаимодействията на протеини с ДНК с висока прецизност.
Изследователите открили повече от 4000 гена, свързани с MECP2. Те повторили картографирането си в неврони с често срещани MECP2 мутации, свързани със синдрома на Рет, за да определят къде MECP2 е изчерпан в болестното състояние.
Познаването на гените, с които MECP2 се свързва, позволи на Лиу и Фламиер да започнат да правят връзки между мишените на MECP2 и здравето на мозъка. Те откриха, че много от неговите мишени участват в развитието и функцията на невронните аксони и синапси.
Те също така сравниха своя списък с MECP2 мишени с базата данни на Simons Foundation Autism Research Initiative (SFARI) за гени, свързани с аутизъм, и установиха, че 381 гена в тази база данни са MECP2 мишени.
Източник: Neuron (2024). DOI: 10.1016/j.neuron.2024.04.007
Тези открития могат да помогнат за изясняване на механизмите, лежащи в основата на симптомите на аутизъм при синдрома на Рет, и да предоставят добра отправна точка за изследване на възможната роля на MECP2 при аутизъм.
„Създадохме първата интегрирана карта на епигенома MECP2 в здравето и болестта и тази карта може да насочи бъдещите изследвания“, казва Лиу. „Познаването на това кои гени са мишени на MECP2 и кои гени са директно нарушени при заболяването, осигурява солидна основа за разбиране на синдрома на Рет и задаване на въпроси относно генната регулация в невроните.“
Изследователите също така са изследвали дали MECP2 увеличава или намалява експресията на целевите си гени. В съответствие с историята на идентифицирането на MECP2 от някои като активатор, а от други като репресор, Лиу и Фламие са открили примери, при които MECP2 е играл и двете роли.
Въпреки че MECP2 по-често се смята за репресор, Лиу и Фламиер откриха, че той е предимно активатор – потвърждавайки предишни открития на Йениш и Лиу. Един нов експеримент показа, че MECP2 активира поне 80% от своите мишени, а друг установи, че активира до 88% от своите мишени.
Картата на целевите гени, създадена от изследователите, предостави допълнителна информация за ролята на MECP2 като активатор. Те откриха, че гените, които MECP2 активира, обикновено се свързват с регион на ДНК преди гена, наречен място за начало на транскрипцията.
Това е мястото, където клетъчният механизъм инициира процеса на транскрипция на ген в РНК, след което РНК се транслира във функционален протеин, който е продукт на генната експресия. Наличието на MECP2 в мястото на начало на транскрипцията, където започва генната експресия, е в съответствие с ролята му на генен активатор.
След това изследователите се заели да определят каква роля играе MECP2 в активирането на гените. Те изследвали с кои молекули се свързва MECP2 на това място, в допълнение към ДНК, и открили, че MECP2 взаимодейства директно с протеинов комплекс, наречен РНК полимераза II (РНК Pol II). РНК Pol II е ключова клетъчна машина, която транскрибира ДНК в РНК. РНК Pol II не може да намира гени самостоятелно, така че се нуждае от различни кофактори или протеинови сътрудници, за да си върши работата.
Изследователите предполагат, че MECP2 служи като един такъв кофактор, помагайки на РНК Pol II да инициира транскрипцията в гените, където MECP2 се свързва. Структурният анализ на MECP2 е идентифицирал части от молекулата, които се свързват с РНК Pol II, а други експерименти са потвърдили, че загубата на MECP2 намалява наличието на РНК Pol II в подходящи места за начало на транскрипцията, както и нивата на експресия на целевите гени.
Това предполага, че синдромът на Рет може да е причинен от намалена транскрипция на гени, към които е насочен MECP2, поради мутации в MECP2, които му пречат да се свърже с РНК Pol II или с ДНК. В съответствие с тази идея, най-често срещаните мутации в MECP2, свързани с болестта, са отрязвания: мутации, при които липсва част от протеина, което може да промени взаимодействието между MECP2 и РНК Pol II.
Изследователите се надяват, че техните открития не само ще променят разбирането ни за MECP2, но и че по-задълбоченото и по-широко разбиране за това как MECP2 влияе върху развитието и функцията на мозъка може да доведе до нови прозрения, които ще помогнат на хората със синдром на Рет и свързани с него разстройства, включително аутизъм.
„Този проект е чудесен пример за съвместния характер на лабораторията „Яниш“, казва Фламиер. „Рудолф и аз имахме специфичен проблем, свързан със синдрома на Рет, а аз имах опит с технологията CUT&Tag, която можеше да реши проблема. Чрез дискусия осъзнахме, че можем да обединим усилията си и сега имаме голямо хранилище с информация за MECP2 и връзките му с болестите.“